
SETD2调控组蛋白甲基转移酶H3K36me3的分子机制及其与肿瘤的关系
2021-03-22 00:30王思思伍彩霞申志华广东医科大学基础医学院病理学系病理生理教研室广东湛江524023
广东医科大学学报 2021年1期
邹 园,王思思,伍彩霞,揭 伟,申志华(广东医科大学基础医学院 .病理学系;2.病理生理教研室,广东湛江 524023)
表观遗传学是指非依赖 DNA序列改变所致的基因表达水平变化,由此引起生物学表型的改变。表观遗传学修饰类型主要包括 DNA甲基化、乙酰化、组蛋白修饰、组蛋白异构体、基因组印记等。DNA甲基化是表观遗传学中最重要的修饰机制之一,其主要发生在基因启动子区域,通过甲基化修饰作用影响染色体结构,从而调控基因转录表达。组蛋白甲基化在真核细胞转录调控中起着重要作用。SET domain containing 2(SETD2)作为人体组织中唯一的组蛋白第36位赖氨酸三甲基转移酶(H3K36me3)的修饰酶,其基因突变在人类肿瘤组织中普遍存在,SETD2蛋白低表达与肿瘤临床进展关系密切。本文旨在从表观遗传学角度总结 SETD2在肿瘤形成和发展过程中的作用及机制,为肿瘤防治提供理论基础。
1 SETD2基因及其编码蛋白质的结构特征
SETD2别名亨廷顿蛋白相互作用蛋白1,最早由人造血干细胞分离。人类SETD2基因位于3 号染色体短臂(3p21.31),长约192 kb,由 23个外显子和22个内含子组成,有 9个转录本(剪切变体),各转录本基本生物学信息见表1。全长SETD2蛋白质由2564个氨基酸组成,分子量为280 KDa。SETD2蛋白包含多个结构域,包括(1)SET(Suvar3-9,Enhancer-of-zeste,Trithorax)域:由 AWS、SET和PS(post-SET)模体组成,该结构域是执行组蛋白 H3第36位赖氨酸单甲基化(H3K36me)转移的催化结构域[1]。(2)H4/H2A相互作用域:由一小段酸性残基组成,其作用可能是稳定核小体上的 SETD2或将酶结构域定位在组蛋白H3第36位赖氨酸(H3K36)残基上[1]。(3)WW 和CC域:两者均为保守区域。WW结构域是富含脯氨酸的蛋白质结合。在细胞核中,RNA聚合酶Ⅱ(RNAPⅡ)的C末端结构域(CTD)会通过与具有该结构域的蛋白质结合调节转录。在某些蛋白质中,CC结构域能够促进同源二聚化。(4)自抑制结构域(AID):位于SET和WW域之间,作用是抑制 SETD2过度活跃,减少核小体和ATM基因上H3K36me的数量并影响SETD2与RNAPII的作用。(5 )Set2-Rpb1相互作用(SRI)域:通过RNAPII的丝氨酸2(Ser2)和丝氨酸5(Ser5)双磷酸化CTD重复序列,与H3K36甲基化一起参与转录的延长[2]。
2 SETD2与组蛋白H3K36me3调控
组蛋白甲基化及其相关酶是染色质结构和基因转录的重要调控因子。SETD2通过多种机制参与组蛋白修饰,组成了复杂的表观遗传转录调控网络。近年来,以组蛋白共价修饰为主要标志的表观遗传调控研究已成为生命科学前沿快速发展的热点领域,其中赖氨酸甲基转移酶成为了调节基因转录或信号通路的新靶点。组蛋白赖氨酸特异性甲基化在DNA的各个方面都发挥着重要作用如转录、DNA复制、重组和DNA损伤修复。SETD2可能通过以下几个方面实现对 H3K36me3的调节:(1 )RNAPII CTD中Ser2 和Ser5磷酸化。CTD磷酸化在 H3K36甲基化的调控中起关键作用。RNAPII 的CTD激酶CTK1(CTD kinase 1)的缺失或CTD的部分缺失,导致H3K36甲基化的选择性废除,表明CTD磷酸化在 H3K36甲基化中的重要作用。其中,SRI结构域与双重修饰的CTD重复序列(Ser2 和Ser5磷酸化)的特异结合具有高亲和力。同时,已经含有磷酸化 Ser2 或 Ser5 的 CTD底物更容易被CTK1磷酸化。但研究发现双磷酸化的Ser2和Ser5同时出现的频率较低,这表明Ser2-CTD磷酸化可能是SETD2相互作用的主要标记[1,3]。(2)组蛋白伴侣 Spt6。Spt6作为一种保守的转录因子和组蛋白伴侣也调节 SETD2介导的 H3K36me3过程。Spt6蛋白包含一个C 端保守的SH2结构,该结构域被招募到RNAPII的磷酸化CTD中,参与转录、mRNA处理和组蛋白修饰的各种因子的招募[4]。H3K36me3需要Spt6、RNAPII的 CTD、CTK1 和SETD2 的 SRI结构域的完整性。而SETD2催化的组蛋白 H3 第36位赖氨酸二甲基化(H3K36me2)在很大程度上独立于 CTK1依赖的 CTD磷酸化和SETD2的 SRI结构域。CTK1和Spt6相互调节彼此的 SETD2蛋白质稳定性。但是,Spt6维持 Ser2-CTD磷酸化水平的能力弱于 Spt6控制H3K36me的能力[1]。(3 )RNA聚合酶Ⅱ相关因子1(Paf1)复合体。Paf1复合物与RNAPII结合,促进转录延长,在转录过程中协调多个组蛋白翻译后修饰[1]。已知Paf1复合体含有Rtf1、Cdc73、Paf1、Ctr9 和Leo1亚基,Paf1复合体组分的缺失对组蛋白H 3 第4 位赖氨酸(H3K4)甲基化水平有不同的影响。Rtf1缺失消除了组蛋白 H3第4位赖氨酸单甲基化(H3K4me)并严重降低了组蛋白 H3 第79位赖氨酸单甲基化(H3K79me),而 Leo1突变不影响 H3K4me 或 H3K79me[5]。研究表明,Paf1C亚单位 Rtf1的组蛋白修饰域(连接 Paf1和RNAPII的 Paf1复合物的一个成员)直接与泛素共轭酶Rad6相互作用并独立于转录刺激 H2Bub[6]。但是,亦有研究表明,Paf1复合物的缺失导致 Ser2-CTD磷酸化水平降低,Paf1的这种作用可能是间接的,且Paf1可能通过与Spt6作用而稳定Spt6,表明Paf1复合物有助于稳定和/或招募Spt6 和CTK1到染色质[7]。一旦加入染色质,CTK1可以磷酸化RNAPII-CTD而结合SETD2[1]。(4)组蛋白 H3K36去甲基化。由于组蛋白甲基化是由组蛋白甲基化酶和去甲基化酶(如LSD1和JHDM1)动态相互调节,因此,组蛋白甲基化也受去甲基化的调节。在芽殖酵母中,有两种主要的H3K36me去甲基酶,如具有 JmjC结构域的蛋白Jhd1和Rph1。Jhd1作用于 H3K36me 和H3K36me2,而Rph1作用于 H3K36me2和H3K36me3。在高等真核生物中,赖氨酸去甲基酶 KDM2A可以去除H3K36的甲基化,其机制主要是 CpG岛直接招募 H3K36特异性 KDM2A,从而产生 CpG岛染色质被耗竭。KDM2A利用锌指CxxC(ZF-cxc)结构域优先识别非甲基化 CpG-DNA。当 CpG-DNA甲基化时,结合被阻断,从而将KDM2A限制在非甲基化CpG岛上而达到去除甲基化的目的。此外,有研究表明KDM2A与异染色质蛋白(HP1)相互作用,将 KDM2A招募到组蛋白 H3第9位赖氨酸三甲基化(H3K9me3)修饰的核小体,介导染色质沉默[8-9]。(5)非编码RNA。在转录前水平,长非编码RNA HOTAIR 通过减少 SETD2启动子区域募集 CREB、P300和RNAPII从而抑制SETD2的表达和磷酸化[10]。有研究表明,miR-106b-5p的过度表达导致透明细胞性肾细胞癌(ccRCC)细胞中SETD2的 mRNA和蛋白水平下降[11]。迄今,有关涉及调节 SETD2水平的其他 RNA因子还有待进一步探索。
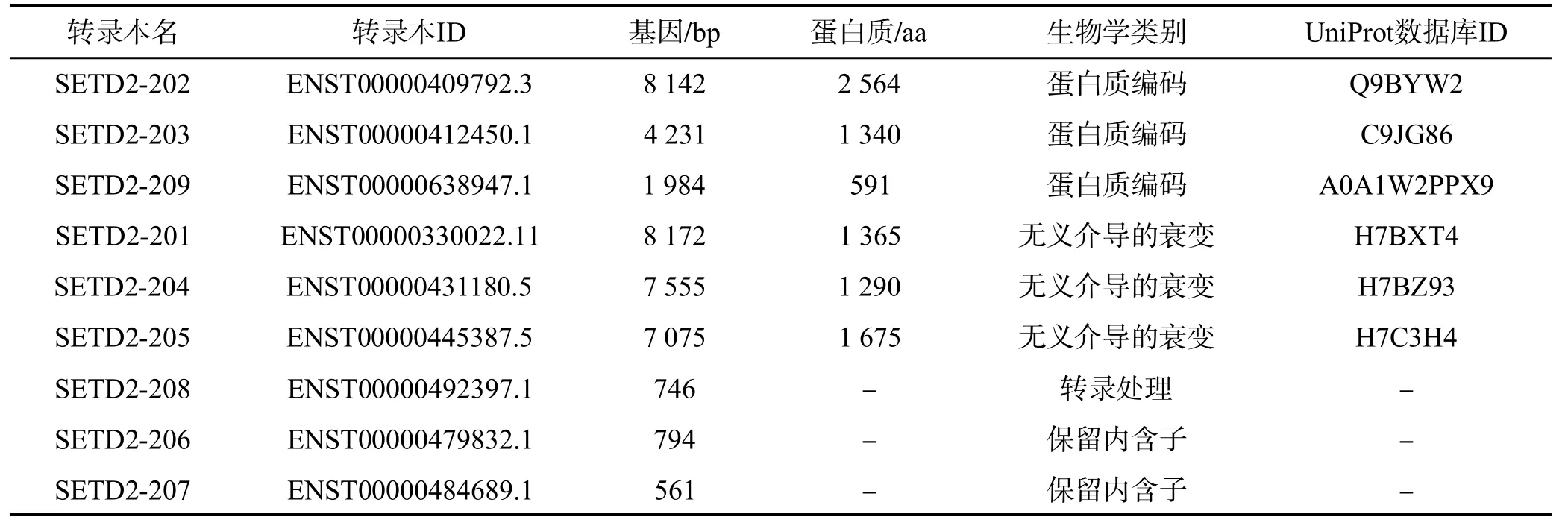
表1 人SETD2基因转录本的基本生物学信息
3 SETD2致癌机制
研究表明 SETD2基因在多种人类肿瘤中普遍存在突变现象,如急性淋巴细胞性白血病、乳腺癌、肾透明细胞癌、结直肠癌和脑胶质细胞瘤等,这可能是众多肿瘤组织中 SETD2蛋白表达水平低下的原因之一。有关 SETD2异常的致癌机制可能与如下几个方面关系密切:(1)SETD2突变后下调H3K36me3表达水平,增加了体内错误的转录起始过程,最终导致基因转录异常。H3K36me3与转录起始的频率相关,保证了转录的高保真度,从而抑制细胞内隐藏的错误转录起始过程。此外,在缺乏H3K36me3的肿瘤中,mRNA处理缺陷(包括内含子保留和异常剪接)影响了基因组中多达25%的基因表达。同时,可以观察到错配的外显子附近的核小体占有率降低。同时,H3K36me3在 S期早期达到峰值,表明SETD2在 DNA复制过程中最活跃。另外,在肾癌中,SETD2基因缺陷导致 DNA甲基化缺失,导致核小体致密性异常降低,从而阻碍复制叉的进展,并导致细胞在 S期 积聚[12]。故 RNA的丰度、稳定性或剪接的改变可引起磷酸化蛋白质组的改变,并破坏正常的细胞信号和细胞周期检查点,导致肿瘤的发生。(2)SETD2水平下降影响了 p53功能。众所周知,p53是重要的肿瘤抑制基因。在多种SETD2突变的肿瘤中均发现p53蛋白和mRNA水平的降低[13]。研究表明,SETD2与p53的转激活域(TAD)相互作用,后者与泛素蛋白连接酶 HDM2结合,抑制肿瘤抑制因子的活性并促进其降解来增加 P53的稳定性[14]。(3)维持基因组稳定和促进 DNA损伤修复。组蛋白修饰在DNA双链断裂(DSB)修复过程中发挥重要作用。H3K36me3通过与MutSα的 PWWP结构域的物理相互作用募集错配识别蛋白 MutSα来复制染色质,证明H3K36me3与MutSα一起参与保护,免受突变,其在外显子和活跃转录区域中比在内含子和非转录区域中共富集得多。相应地,消耗 H3K36me3或破坏H3K36me3 与MutSα相互作用提高了活跃转录基因中的自发突变频率[15]。此外,SETD2功能缺陷肿瘤细胞通常会出现基因组微卫星不稳定性(MSI),而基因组MSI与肿瘤发生、发展相关[16]。
4 常见肿瘤中SETD2的表达及作用
4.1 泌尿系统肿瘤
肾透明细胞癌(ccRCC)是最常见的成人肾癌。突变、甲基化失活或 von Hippel-Lindau的异常表达是ccRCC发生的主要原因[12]。多项独立研究对ccRCC临床样本进行了蛋白编码基因和外显子测序,同样发现了 SETD2的失活突变。肾癌中的 SETD2不仅在表观遗传功能障碍中发挥功能,同样在细胞代谢调节中起作用[17]。表观遗传学修饰方面,H3K36me3可与H4K16ac协同促进转录激活,而 SEDT2缺失导致 H3K36me3的蛋白水平明显下调[18],影响基因的甲基化修饰。ccRCC也是一种高度“代谢重编程”疾病,能量、营养和氧感应等方面均存在异常[19]。SETD2的缺失可下调肌酸、糖胺聚糖和碳水化合物的代谢过程,通过 PGC1α介导的代谢网络增强氧化磷酸化和脂质生成[18,20]。这种代谢异常为肾细胞癌的影像学和治疗干预新靶点的开发提供了新的机会。
众所周知,microRNA在转录后水平调节基因的表达,其异常在肿瘤发生中的起重要作用。在ccRCC组织和细胞系中,内源性 miR-23b-5p、miR-34b-3p和miR-106b-5p的高表达与SETD2低水平相关,其中miR-106b-5p可直接靶向结合SETD2 mRNA的3'UTR,提示 microRNA可能直接调控 SETD2的表达[21-22]。尽管人们已经认识到 SETD2在调节ccRCC细胞的增殖和凋亡中发挥重要作用,但迄今SETD2在ccRCC中的失活机制仍有待进一步阐明。
4.2 淋巴造血系统肿瘤
以染色体重排为特征的急性白血病需要额外的分子学异常刺激才能发展成完全的恶性肿瘤,然而其协同机制仍不清楚。SETD2在血液恶性肿瘤的发生和治疗敏感性中起重要作用。混合系白血病(MLL)基因编码一种DNA结合蛋白,介导H3K4的甲基化。MLL易位后编码一种 MLL融合蛋白,失去了H3K4甲基转移酶调节活性,却能有效地将造血细胞转化为白血病干细胞。在MLL重排白血病和复发性急性白血病中常发生SETD2基因突变,SETD2突变的白血病细胞的细胞周期进程、S 和G2/M检查点调控相关的信号减弱,进而影响DNA复制、有丝分裂和细胞死亡[23]。研究发现,SETD2基因敲除后导致伊马替尼不敏感和白血病干细胞在慢性髓细胞白血病(CML)细胞系中富集[24]。最新的报道显示在MLLAF9 AML小鼠模型中使用条件性SETD2基因敲除,纯合性SETD2缺失导致MLL-AF9诱导的白血病发生明显延迟,而杂合子 SETD2缺失则加速疾病发展和化疗抵抗[25]。总之,目前对 SETD2在血液性恶性肿瘤中的发病机制尚不清楚。
4.3 呼吸系统肿瘤
肺腺癌中SETD2基因突变较常见。在肺癌中的H3K36me3丢失可破坏染色质的多样性,如基因剪接、DNA修复、转录伸长、DNA甲基化和基因组完整性的维护。SETD2在肺癌的治疗中也起着关键作用。顺铂是治疗非小细胞肺癌(NSCLC)最常用的化疗药物之一。抑制SEDT2表达和SETD2突变可通过抑制 NSCLC细胞中 H3K36me3 和ERK的激活而产生顺铂耐药性。故SETD2可能在肺癌的发生发展中起到抑癌基因的作用[26]。
报道显示,与鼻咽部炎症组织相比,鼻咽癌组织中SETD2 mRNA水平显著下降[27]。本实验室也发现,鼻咽癌细胞中 SETD2基因敲除后影响了 20多条与肿瘤密切相关的经典信号通路[28]。由此可知,SETD2在鼻咽癌的发生、发展中发挥重要作用。迄今,有关 SEDT2在鼻咽癌中的研究还处在起步阶段,今后仍需进一步探讨SETD2在鼻咽癌的作用及机制,为鼻咽癌的治疗和预后判断提供更多的参考信息。
4.4 神经系统肿瘤
恶性原发性中枢神经系统肿瘤是儿童癌症相关死亡的主要原因。儿童神经胶质瘤中发现SETD2突变率约15%,而成人神经胶质瘤中 SETD2突变率为8%[29]。SETD2的失功能突变在小儿及青年人的晚期神经胶质瘤中可以被发现,但早期神经胶质瘤中却并未检测到,这代表SETD2的失功能突变在神经胶质瘤中具有晚期特异性。SETD2失活会导致胶质瘤进展,并向高临床分期演进。
4.5 骨肿瘤
对骨肉瘤易感犬的外显子测序发现,骨肉瘤肿瘤表现出高频率体细胞拷贝数改变。Gardner等[30]研究发现21%的病例中发生了 SETD2突变,其突变类型包含了点突变、移码插入或删除等。在对 24名脊索瘤患者的分析中,SETD2突变主要是点突变或拷贝数丢失[31]。有关SETD2与骨肉瘤生物学功能的报道目前尚缺乏。
4.6 消化系统肿瘤
随着 SETD2表达的增加,胃癌细胞 HGC-27和AGS的迁移、增殖和侵袭能力下降,即SETD2的低表达与胃癌不良预后正相关[32]。SETD2基因失活使小鼠肠上皮促进肠干或祖细胞的自我更新和组织再生[33]。
4.7 其他系统肿瘤
根据 TCGA和代谢数据库,SETD2在乳腺癌各亚型中均有突变。在所有类型的乳腺癌中,SETD2的表达水平与患者的预后显著正相关[17,34]。SETD2的表达与乳腺癌分级、分期及淋巴结转移呈负相关,表明SETD2可能在肿瘤抑制中起作用。由于其与p 53异常活性相关,因此可以作为乳腺癌的预后标志物。
SETD2基因在多种人类肿瘤中都存在突变现象,对肿瘤的发生、发展及预后起着重要的作用,这为临床防治提供了相应的理论基础。
5 问题与展望
SETD2基因在多种人类肿瘤中失活,并参与肿瘤的发生与临床进展。基于 SETD2为靶点的表观遗传学研究目前正受到研究人员的重视。今后可在如下几个方面深入研究:(1)明确 SETD2在多种肿瘤组织中呈现的突变现象及其规律,深入阐明常见肿瘤组织中 SETD2的突变热点。分析常见肿瘤组织中SETD2突变与SETD2表达之间的关系。(2)充分阐明SETD2的病理生理作用。基于现有的基因组编辑技术,有望获得SETD2敲除或过表达动物模型,以这些动物模型为对象,分析SETD2表达异常与组织器官发育、分化及系统性疾病的关系。(3)SETD2可作为非组蛋白甲基转移酶,而非组蛋白的甲基化在肿瘤发生、发展中同样发挥重要作用。因此,有必要明确 SETD2对下游非组蛋白基因的甲基化修饰情况,从而有望发现新的致病机制。(4)深化基于SETD2靶点的药物开发。2014年,Cancer Discovery杂志在“研究观察”专栏中点评“SETD2是协同血液肿瘤发生和维持中的抑癌基因”的重要影响,因此SETD2作为一个新的分子治疗靶点,已经在急性白血病的诊断和治疗等提供了新的机遇。其中,应用高通量技术为 SETD2作用相关的药物筛查提供了思路。
综上,SETD2是一个有巨大潜力的肿瘤分子治疗靶点,深入研究 SETD2在肿瘤形成和发展过程中的作用和机制,对于肿瘤的诊断、治疗和预防具有重要意义。同时期待随着表观遗传学的深入研究,对SETD2的表达调控通路作出新的解释。
猜你喜欢
河北果树(2021年4期)2021-12-02
上海公路(2019年3期)2019-11-25
福建基础教育研究(2019年10期)2019-05-28
广东饲料(2016年3期)2016-12-01
动物营养学报(2015年10期)2015-12-01
癌变·畸变·突变(2015年3期)2015-02-27
现代检验医学杂志(2015年2期)2015-02-06
应用化工(2014年10期)2014-08-16
遗传(2014年3期)2014-02-28
遗传(2014年3期)2014-02-28
