
Drug target discovery by magnetic nanoparticles coupled mass spectrometry
2021-03-22 02:38DndnXiBolingLiuXioweiXuDingQiulingZheng
Dndn Xi , Boling Liu , Xiowei Xu , Y Ding ,*, Qiuling Zheng ,**
a Department of Pharmaceutical Analysis, School of Pharmacy, China Pharmaceutical University, Nanjing, 210009, China
b State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing, 210009, China
c Key Laboratory of Drug Metabolism and Pharmacokinetics, China Pharmaceutical University, Nanjing, 210009, China
Keywords:Drug-target discovery Nanoparticle Mass spectrometry
ABSTRACT Drug target discovery is the basis of drug screening.It elucidates the cause of disease and the mechanism of drug action, which is the essential of drug innovation. Target discovery performed in biological systems is complicated as proteins are in low abundance and endogenous compounds may interfere with drug binding. Therefore, methods to track drug-target interactions in biological matrices are urgently required.In this work, a Fe3O4 nanoparticle-based approach was developed for drug-target screening in biofluids. A known ligand-protein complex was selected as a principle-to-proof example to validate the feasibility. After incubation in cell lysates, ligand-modified Fe3O4 nanoparticles bound to the target protein and formed complexes that were separated from the lysates by a magnet for further analysis.The large surface-to-volume ratio of the nanoparticles provides more active sites for the modification of chemical drugs. It enhances the opportunity for ligand-protein interactions, which is beneficial for capturing target proteins, especially for those with low abundance. Additionally, a one-step magnetic separation simplifies the pre-processing of ligand-protein complexes, so it effectively reduces the endogenous interference. Therefore, the present nanoparticle-based approach has the potential to be used for drug target screening in biological systems.
1. Introduction
The identification of the protein targets of small molecule drugs is of great significance and is currently an unmet challenge. The ability to discover new drug targets in biological systems is crucial for pharmaceutical research and essential for gaining a deep understanding of disease mechanisms. With the completion of the Human Genome Project, it is estimated that the drug target pool contains at least 1,800 proteins,but only a few are currently under investigation for innovative drug development [1]. Therefore,sensitive and unambiguous identification of drug target proteins is the basis of drug screening as well as mechanistic studies and subsequent optimization of the drug structure.
Conventional methods,such as nuclear magnetic resonance [2]and X-ray crystallography [3], provide high spatial resolution investigation of small molecule-target interactions;however,their usefulness is limited by the sample purity and amount requirements.Microscale thermophoresis is used for binding affinity measurement (Kdvalue) [4], but requires a recombinant protein with a fluorescent label. Mass spectrometry (MS) is an alternative method to measure small molecule-target interactions and has the benefits of relatively high selectivity and sensitivity [5-7]. The introduction of mild ionization methods, such as desorption electrospray ionization (DESI) [8,9] and native MS [10,11], maintains non-covalent interactions and enables the direct detection of compact small molecule-target complexes. Complementary information, such as binding specificity, binding site identification and conformational changes,can be provided by coupling with selected analytical techniques together[5,7,12].However,the applications of these methods to target discovery are indirect. They mainly focus on the binding characteristic description of identified interactions rather than unknown target screening. Additionally, target discovery performed in biological systems increases in difficulty as proteins are in low abundance and endogenous molecules compete with target binding. Thus, the identification of potential targets along with the exclusion of false-positive results is a challenging task. Therefore, the specificity of screening and identification is a necessary foundational step for drug-target discovery from biological matrices.
Activity-based protein profiling (ABPP) is one of proteomic approaches that have been widely used for drug target discovery.Fluorescent or affinity probes are designed based on the structure of small molecule drugs. Once they are bound with their potential targets, they are isolated by affinity enrichment or gel electrophoresis for further identification [13-15]. Alternatively, immobilization of the small molecule drug on a stationary phase[16,17]can be performed to enrich targets with low abundance and improve detection sensitivity. Such chemical modification-based strategies greatly enhance the detection sensitivity for targets with low abundance; however, the bioactivity after modification should be carefully validated. Methods including cellular thermal shift assay[18,19],drug affinity responsive target stability[20,21]and limited proteolysis-MS [22,23] have been developed to overcome these limitations and are considered to be more universally applicable approaches to drug-target discovery. Potential protein targets are filtered according to their susceptibility changes (stability or conformational changes) to thermo or protease hydrolysis upon binding.However,there are still some targets that may be missed if they have no significant changes after binding or have low binding affinity to a drug. Recently, nanoparticles (NPs), novel carriers for small molecule drugs, have found wide applications in drug delivery and distribution imaging, promoting drug solubility,bioavailability, and stability while reducing dosage and administration frequency[24-27].Their relatively large surface-to-volume ratio,especially compared with that of micron-scaled particle sized agarose beads, greatly increases the loading capacity [27-29].
Herein, a Fe3O4NP-based strategy was developed for small molecule-protein discovery in biological systems (hereafter referred to as ligand-protein). According to the designed strategy,Fe3O4NPs were first made hydrophilic by dopamine modification so that they would be sufficiently dispersed in biofluids. The exposed amino groups offered possible reaction positions for carboxyl group containing ligands.On this basis,the large surfaceto-volume ratio of the NPs promoted ligand loading capacity,which increased the possibility of target interactions [26]. Upon incubation,the formed protein-ligand-NP complexes underwent one-step magnetic isolation from the matrix to reduce sample complexity.The captured proteins were subsequently released through denaturation and subjected to enzymatic digestion prior to MS analysis.Results suggested that target proteins with low abundance were successfully captured from the biological matrices by the designed strategy, making it useful for high-throughput drug target discovery and associated mechanistic studies.
2. Materials and methods
2.1. Chemicals and reagents
DL-dithiothreitol (DTT), iodoacetamide (IAA), formic acid (FA),trypsin,1-ethyl-3-(3-dimethyl aminopropyl)carbodiimide(EDC),Nhydroxysuccinimide (NHS), dexamethasone and McCoy’s 5A medium were all purchased from Sigma Aldrich(St Louis,USA).DMEM/F12 medium, mammalian protein extraction reagent, and insulintransferrin-selenium solution were purchased from Thermo Fisher Scientific (Waltham, USA). Dopamine was purchased from Aladdin(California, USA). Ethanol was purchased from Titan Scientific Co.(Shanghai,China).Fetal bovine serum was purchased from Biological Industry (Kibbutz, Israel). Bicinchoninic acid (BCA) assay kit was purchased from Beyotime(Nantong,China). Obeticholic acid(OCA),GW4064, and tropifexor (LJN452) were all purchased from Med-ChemExpress (New Jersey, USA). Ammonium bicarbonate, urea,FeCl3⋅6H2O, trisodium citrate monohydrate, ethylene glycol, and sodium acetate were all purchased from Sinopharm Chemical Reagent Co. (Shanghai, China). Acetonitrile (ACN, LC-MS grade) was purchased from Merck (Darmstadt, Germany),and deionized water was prepared using a Milli-Q system (Millipore, Billerica, USA). The 15 nm amide iron oxide nanoparticles were purchased from Ocean Nonotech (San Diego, USA). The human colorectal cancer cell line HCT116 and alpha mouse liver 12 cell line AML12 were purchased from the ATCC(Manassas,VA,USA).
2.2. Synthesis of Fe3O4 NPs
The carboxylate functionalized Fe3O4NPs were prepared through a modified solvo thermal reaction[30,31].Typically,1.3 g of FeCl3⋅6H2O and 0.5 g of trisodium citrate monohydrate were first dissolved in 40 mL of ethylene glycol. Afterward, 2.6 g of sodium acetate was added with stirring.The mixture was stirred vigorously for 30 min and then sealed in a teflon-lined stainless-steel autoclave (100 mL capacity). The autoclave was heated at 200°C and maintained for 8 h,and then allowed to cool at room temperature.Some portion of the products was dispersed in water and excess dopamine was added with stirring over several hours. The black products were magnetically separated and washed with ethanol and water 5 times and dried at 60°C for 12 h.
2.3. Ligand modification process
A total of 1 mg of EDC was added to 90 μL of Fe3O4NPs solution(10 mg/mL),vortexed,and then 1.5 mg of NHS was added to initiate the condensation reaction.The obtained mixture was incubated at 65 rpm, 37°C for 15 min. A portion of 10 μL of ligand (OCA/GW4064/LJN452, 10 mM/L) was added and incubated at 65 rpm,37°C for 10 h.The obtained modified NPs were stored at 4°C in the dark before use.
2.4. Nano liquid chromatography-MS analysis
Details of protein sample preparation could be found in the Supplementary Material. Peptide mixtures generated from enzymatic digestion were subjected to chromatographic separation using a nanoAcquity UPLC system(Waters,Milford,USA)equipped with a Waters Acquity M-Class UPLC HSS T3 column(75 μm×150 mm,1.8 μm).The mobile phase consisting of 0.1%FA in H2O (phase A) and 0.1% FA in ACN (phase B) was delivered at a flow rate of 0.3 μL/min following a gradient program of 1%(B)from 0 to 1 min,1%-40% (B) from 1 to 95 min, 40%-85% (B) from 95 to 96 min,85%(B)from 96 to 99 min,85%-1%(B)from 99 to 100 min,and 1%(B)from 100 to 120 min.The LC eluent was analyzed using a Waters G2-Si Q-TOF mass spectrometer(Waters,Milford,USA).The source capillary voltage was set at 3000 V, and the source temperature was 80°C.The scan time of the MS was set as 0.2 s,and the full MS scan range was set as 350-2000 m/z with a scan time of 0.2 s. The MS/MS scan range was set as 50-2000 m/z. The top 10 abundant precursors were subjected to MS/MS fragmentation with a ramp collision energy set between low energy (14-19 eV) and elevated energy(67-94 eV)using a scan time of 0.1 s per function.
3. Results and discussion
Obeticholic acid(OCA) and the ligand-binding domain(LBD) of its target protein Farnesoid X Receptor (FXR) were selected as a principle-to-proof example to explore the feasibility of the designed Fe3O4NP-based strategy. Fe3O4NPs of 100 nm size(Figs.1A and B)were chosen because of their remarkable magnetic properties, biocompatibility and stability. Fe3O4NPs were first made hydrophilic by dopamine modification on the surface. Besides being compatible with biofluids, the exposed amino groups from the dopamine also afforded reaction sites for the carboxylcontaining OCA (the OCA structure is shown in Fig.1C). The OCAmodified NPs (referred to as OCA-NP) were obtained through a condensation reaction and were confirmed by UV detection(details found in the Supplementary Material). As shown in Fig.1D, OCANPs displayed a maximum absorption around 206 nm that was comparable to that of OCA, while no obvious UV absorption was observed for the NPs alone. Cell lysate was chosen as a matrix to mimic a complicated biological background (details found in the Supplementary Material)and the LBD was doped at a mass ratio of 1:13(LBD:total protein=1:13).As shown in Scheme 1,the obtained OCA-NPs were first dispersed into the LBD-containing cell lysate.They immediately became homogenous allowing the target interactions to be initiated. The incubation time was 150 min to ensure that OCA made full contact with its target. The resulting target-OCA-NPs were magnetically collected, and the supernatant was removed and maintained for further analysis. The separated target-OCA-NPs were subsequently treated with 8 M urea to disrupt non-covalent bindings to release proteins, which then underwent enzymatic digestion and LC-MS analysis. Similarly, NPs alone were treated with the same procedure and incubated with the same volume of cell lysate so that non-specific protein interactions could be considered. Therefore, a total of four samples were prepared for LC-MS analysis: the supernatant removed from the OCA-NP treated sample (OCA-NP-S), the proteins decomposed from the OCA-NPs(OCA-NP-P),the supernatant removed from the NP treated sample(NP-S),and the proteins decomposed from the NPs (NP-P).
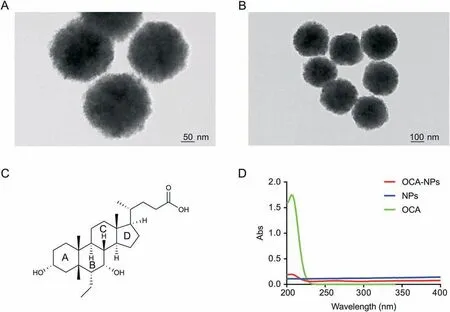
Fig.1. TEM images of 100 nm nanoparticles(NPs)with(A)50 nm scale and(B)100 nm scale;(C)chemical structure of obeticholic acid(OCA);(D)UV absorption of OCA,NPs and OCA-NPs (100 nm).
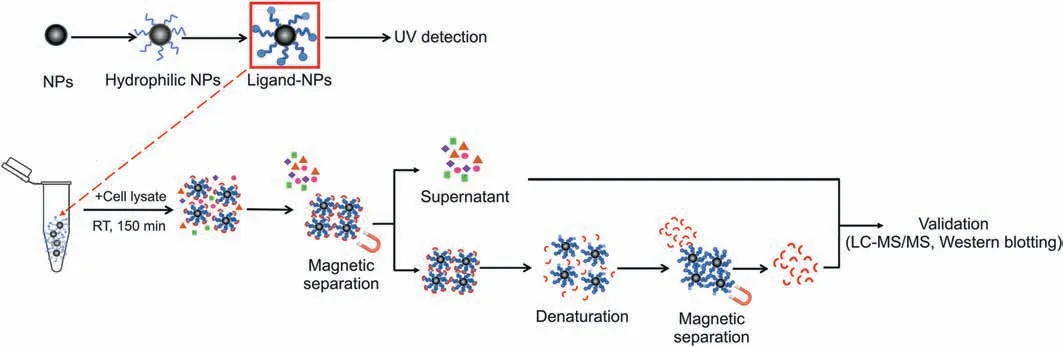
Scheme 1. Ligand-target discovery by designed Fe3O4 NP-based strategy.NP:nanoparticle;UV:ultraviolet;RT:room temperature;lC-MS/MS:liquid chromatography-tandem mass spectrometry.
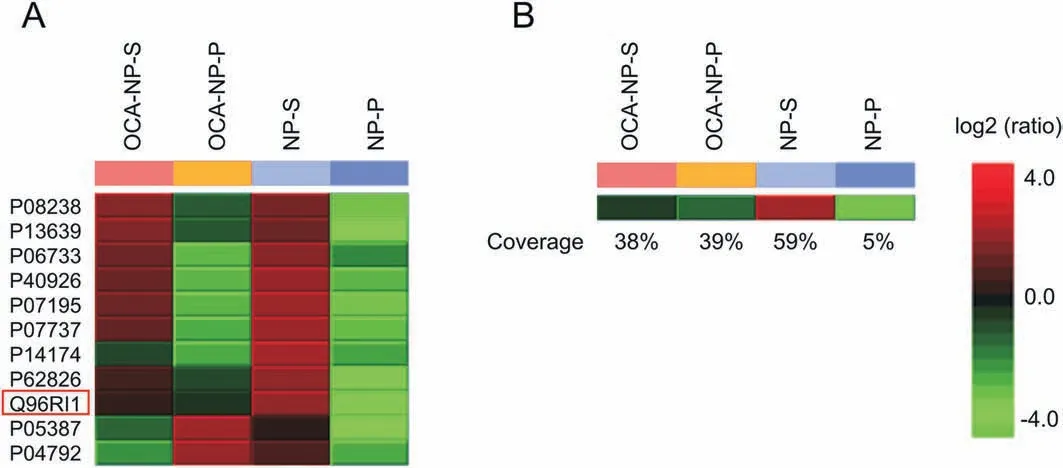
Fig. 2. (A) Identification and relative quantification of proteins obtained from supernatant or decomposed from OCA-NPs/NPs; and (B) LBD sequence coverage results of each sample. OCA-NP-S: supernatant of OCA-NP treated cell lysate; OCA-NP-P: proteins decomposed from OCA-NPs; NP-S: supernatant from NP treated cell lysate; NP-P: proteins decomposed from NPs; OCA: obeticholic acid; NP: nanoparticle.
After LC-MS analysis, the acquired data were interpreted by PEAKS for protein identification and relative quantification(details found in the Supplementary Material).For the NP-P sample,only a few proteins were identified, which were likely captured through non-specific interactions. The proteins that decomposed from the OCA-NPs were identified and potential targets were refined by exclusion of those proteins believed to be acting non-specifically(shown in Fig. 2A). Among the target candidates, proteins with high abundance in the cell lysate,such as profilin-1(ID P07737)and alpha-enolase (ID P06733) (details found in Table S1 in the Supplementary Material),were eliminated and the LBD(ID Q96RI1)became prominent.The LBD was also detected from OCA-NP-S and NP-S samples. Interestingly, the results showed that the NP-S sample contained the highest quantity of the LBD, while OCA-NPP and OCA-NP-S contained lower but comparable amounts. The relative quantification results indicated that the OCA-NPs were selective toward the LBD such that a significant amount was captured and isolated from the cell lysate.No detection of LBD from NP-P sample also confirmed that its capture was due to the OCA modification but not through non-specific interactions. Note that LBD still remained in the supernatant after the OCA-NPs were treated and could be detected (OCA-NP-S sample in Fig. 2A). This may be explained by the OCA modification efficiency, interruption of non-covalent bindings during magnetic isolation,or the fact that the amount of doped LBD was much higher than that in a real biological system. Additionally, the sequence coverage of the LBD(Fig. 2B)from the OCA-NP-P sample (39%)was comparable to that of NP-S(59%),indicating that the LBD captured by the OCA-NPs was enough for MS identification.
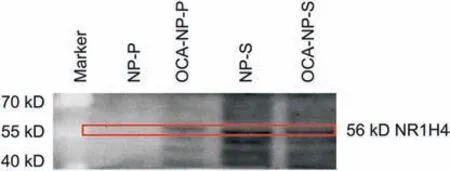
Fig. 3. Immunoblotting of proteins obtained from supernatant or decomposed from OCA-NPs/NPs (15 nm). NP-P: proteins decomposed from NPs; OCA-NP-P: proteins decomposed from OCA-NPs;NP-S:supernatant from NP treated cell lysate;OCA-NP-S:supernatant of OCA-NP treated cell lysate; OCA: obeticholic acid; NP: nanoparticle.
Based on the feasibility investigation using the OCA-LBD complex, two additional FXR ligands, GW4064 and LJN452, were selected to validate the generality of the proposed strategy. Either GW4064 or LJN452 contains one carboxyl group(structures shown in Figs. S1A and B) that can react with the NPs via a condensation reaction.The LBD doped cell lysate was mixed with the ligand-NPs to initiate target binding. The captured proteins were then dissociated by denaturation and subjected to enzymatic digestion prior to MS analysis. Potential targets were identified and refined following the procedure described above.Figs.S1C and D show that the LBD was listed as a candidate target for both ligand-NP-P samples as expected (Tables S2) in significant quantities compared with that from the NP-P sample, especially in the GW4064-NP-P sample. The fact that the amount of the LBD captured by GW4064 was higher than that by LJN452 might be attributed to different modification efficiencies among the two ligands. The results showed that the developed NP-based target discovery strategy was suitable to enable ligand release and allow isolation and recovery of the target in a sufficient amount for identification and relative quantification from a biological matrix.
Based on the verification in the mimic system,it was of interest to determine whether the developed methodology could be applied to target screening in real biofluids.Initially 100 nm Fe3O4NPs were selected for the study;however,only a few proteins were detected under this condition (data not shown). It was likely that ligand loading capacity was still limited,especially considering the challenge of low target protein abundance in biological system. To overcome this limitation and enhance the interactions, increasing the surface-to-volume ratio by using a reduced particle size may be helpful. Thus, 15 nm Fe3O4NPs were selected to increase the surface-to-volume ratio and the number of reaction sites for the ligand.OCA was still used as the exemplary ligand and FXR was the expected target. The modification was first verified by observing a characteristic peak generated at 206 nm in the UV spectrum (Fig.S2). The obtained OCA-NPs were added to cell lysate to examine target binding, and then magnetically collected to remove the unbound content.Non-specific binding by NPs alone was also carried out as a control. As demonstrated herein with the same known ligand-protein system (OCA-FXR complex), the decomposed protein mixture was verified by Western blotting(details found in the Supplementary Material). Fig. 3 clearly shows that the FXR (Gene ID: NR1H4) response appeared in samples OCA-NP-S, OCA-NP-P,and NP-S. The NP-S sample showed the highest amount of FXR while OCA-NP-P and OCA-NP-S had a comparable but lower amounts of FXR. No FXR was detected in the NP-P sample, which further confirmed that FXR did not non-specifically interact with the NPs and only became detectable when the OCA was attached.The remaining portion of the FXR detected from the OCA-NP-S suggested that either the quantity of modified NPs or the modification efficiency could be optimized for detecting trace amount of FXR. It was important to note that no internal standard, such as GADPH, was used in the Western blotting experiment. This was because the sample pretreatment by NPs resulted in the composition and quantity of proteins in each sample being different and thus the sample content could not be normalized.
4. Conclusions
In this study,we demonstrated an NP-based strategy for ligandprotein screening in a cell lysate.The large surface-to-volume ratio of the NPs enabled the improvement of the ligand modification capacity. Additionally, the hydrophilic nature of the NPs helped them to be adequately dispersed in the cell lysate and made full contact with the target proteins. Therefore, proteins with low abundance could be sensed. Moreover, the magnetic property of the NPs allowed them to be isolated in one-step and enriched while maintaining ligand-protein non-covalent bindings. The bound proteins were then released through denaturation, which was compatible with the downstream enzymatic digestion and MS analysis steps.The designed strategy enabled the selective capture of target proteins among biological matrices while effectively eliminating the interference of endogenous proteins and small molecules, which would complicate the detection processes.Nevertheless, the developed method was limited to carboxyl group-containing compounds as the amino group was the only explored reaction site. More functional Fe3O4NPs may be developed in the future to cover a broader range of ligands and drugs.Additionally,the binding efficiency could be further improved as a portion of the target protein was remained in the supernatant.Furthermore,it was also notably that in our results,a few proteins showed high quantities after ligand-NP treatment besides the desired target which might be caused by the relatively low selectivity of proteins or strong non-specific interactions with ligand.Therefore, for screening of unknown targets in biological systems,the subsequent specific interaction differentiation and biological function validation are still necessary and essential.
Declaration of competing interest
Authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81720108032, 81930109,81421005, 81703471, and 31870946), the Natural Science Foundation of Jiangsu Province (Grant No. BK20170740), the 111 Project(Grant No. G20582017001), projects for Major New Drug Innovation and Development (Grant Nos. 2018ZX09711001-002-003 and 2018ZX09711002-001-004), the State Key Laboratory of Natural Medicines at China Pharmaceutical University (Grant No.SKLNMZZCX201817), a “Double-First Rate” project (Grant No.CPU2018GF09), and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2020.02.002.
 Journal of Pharmaceutical Analysis2021年1期
Journal of Pharmaceutical Analysis2021年1期
- Journal of Pharmaceutical Analysis的其它文章
- Comprehensive metabolic profiling of Alismatis Rhizoma triterpenes in rats based on characteristic ions and a triterpene database
- Evaluation of apoptotic effects of mPEG-b-PLGA coated iron oxide nanoparticles as a eupatorin carrier on DU-145 and LNCaP human prostate cancer cell lines
- Development of an analytical method for multi-residue quantification of 18 anthelmintics in various animal-based food products using liquid chromatography-tandem mass spectrometry
- Analytical methodologies for sensing catechol-O-methyltransferase activity and their applications
- Applications and challenges of low temperature plasma in pharmaceutical field
- Solid phase microextraction chemical biopsy tool for monitoring of doxorubicin residue during in vivo lung chemo-perfusion