
Natural compounds as potential inhibitors of SARS-CoV-2 main protease: An insilico study
2021-03-19 01:25AmareshMishraYaminiPathakAnujKumarSurabhiKirtiMishraVishwasTripathi
Amaresh Mishra, Yamini Pathak, Anuj Kumar, Surabhi Kirti Mishra, Vishwas Tripathi✉
1School of Biotechnology, Gautam Buddha University, Greater Noida-201310, India
2Bioinformatics Laboratory, Uttarakhand Council for Biotechnology, Biotech Bhawan, Pantnagar, U.S. Nagar, Uttarakhand-263145, India
3Advanced Centre for Computational and Applied Biotechnology, Uttarakhand Council for Biotechnology, Dehradun-248007, India
4School of Biotechnology, Jawaharlal Nehru University, New Delhi, India
ABSTRACT
KEYWORDS: SARS-CoV-2; COVID-19; Amentoflavone;Guggulsterone; Puerarin; Piperine
1. Introduction
Coronavirus disease (COVID-19) is an infectious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) which primarily affects the lungs and shows certain types of pneumonia-like symptoms[1]. SARS-CoV-2 is a novel strain of coronavirus and has expanded all over the world in a very short period[2]. The outbreak was declared as a Public Health Emergency of International Concern by WHO on 30 January 2020[3]. To date, there is no specific treatment for this ongoing COVID-19 pandemic. Some preliminary study results investigated the effect of a potential combination of lopinavir and ritonavir on COVID-19 infected patients, which were earlier used in human immunodeficiency virus and SARS CoV or Middle East respiratory syndrome (MERS) coronavirus patients[4,5]. These drugs can specifically target the virus replication cycle. Effective antiviral therapies are still urgently required due to subsequent infection[6].Because of enormous structural and chemical diversity, availability of more chiral centers, and relative biosafety, natural compounds are considered as an excellent source of drugs for several diseases including viral infections. Around 45% of today's best selling drugs have either originated from natural products or their derivatives[7].Natural compounds possess antiviral property and could become a valuable resource. Jin et al. have revealed the crystal structure of SARS-CoV-2/COVID-19 main protease (M/3CL protease, PDB ID-6LU7)[8]which has been structured and repositioned in the Protein Data Bank (PDB) and is publicly accessible. Mof SARS-CoV-2 is reported to play a vital role in virus replication and transcription, which suggests that it could be a promising target for inhibition of the SARSCoV-2 cycle[9].
In the current study, we have selected several natural compounds based on an extensive literature search of natural compounds with anti-viral property. These natural compounds are presented in Supplementary Table 1. We screened and explored the potential of selected natural compounds to inhibit the Mof COVID-19 using molecular docking, followed by molecular dynamic (MD) simulations,molecular mechanic Poisson-Boltzmann surface area (MM/PBSA) free energy calculations, absorption, distribution, metabolism, and excretion(ADME) screening, drug-likeness, target-specific binding, and toxicity analysis.
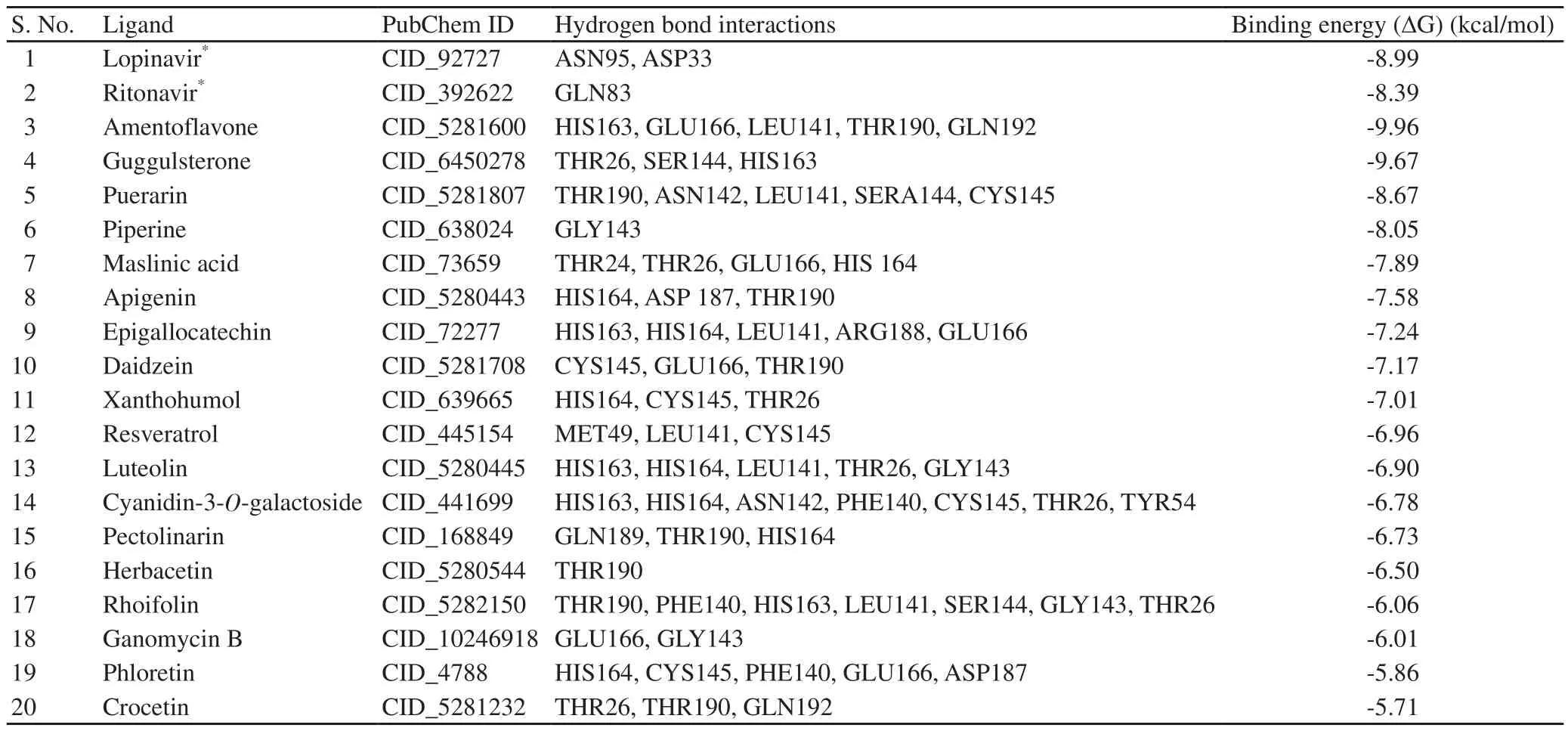
Table 1. Molecular docking analysis results of selected natural compounds against SARS-CoV-2 main protease Mpro (PDB-6LU7).
2. Materials and methods
A flow chart of pipeline of this study is summarized in Supplementary Figure 1.

Figure 1. Docking analysis visualization of SARS-CoV-2 main protease Mpro (PDB-6LU7) binding with lopinavir, ritonavir, amentoflavone, and guggulsterone. The 3D structures of protein-ligand interactions were visualized by discovery studio programs. The binding residues and their chains were identified from the protein-ligand complex.
2.1. Literature survey and ligands selection
An extensive literature survey was conducted to select natural compounds with antiviral properties from different medicinal plants using PubMed and Google scholar platforms. Based on the literature survey, a total of twenty natural compounds were selected, and their chemical structures were extracted from PubChem[10]repository in SDF format. To prepare the ligands to perform molecular docking,hydrogen atoms were added followed by PDB structure generation by Open Babel program. Further, all the molecules were subjected to the energy minimization and optimization using the universal force field at 200 descent steepest algorithm of Open Babel available in PyRx (https://pyrx.sourceforge.io/) and converted in .pdbqt format.
2.2. Preparation of protease
The 3D coordinates of the main Mof SARS-CoV-2 were obtained from the RCSBPDB repository with PDB ID 6LU7[8]. To prepare the macromolecule for docking, water, and other nonspecific molecules were removed by using UCSF Chimera[11]. Polar hydrogen atoms were added to the 3D structure model of Mto allow intramolecular interactions through hydrogen bonds. The structure optimization and energy minimization were performed by using the SPDB viewer[12].While clean geometry module embedded in the Discovery Studio package was utilized for the side chain angles correction.
2.3. Molecular docking
To identify new potential inhibitors against the Mof SARSCoV-2, the site-specific docking-screening of all selected natural compounds were carried out by AutoDock4.2[13]. The box dimensions were kept as 70 Å×70 Å×70 Å with a total of 50 genetic algorithm run. Other docking parameters were set as default.During molecular docking, the amino acid residues including Thr25,Thr26, Gly143, Ser144, His163, His164, and Glu166 were utilized as the binding pocket sites. The protein-ligand interactions were further calculated by the Maestro and Discovery studio programs.
2.4. ADME screening
An in-silico tool for analysis of ADME was used to screen the above-mentioned compounds which could be bioactive via oral administration. Drug-like properties were calculated based on Lipinski's rule of five using SwissADME prediction (http://www.swissadme.ch/)[14]. Canonical SMILES from PubChem was used to identify ADME properties by SwissADME. Various parameters of compounds were analyzed like lipophilicity, molecular weight,hydrogen-bond donors, hydrogen-bond acceptors, clog P-value,Ghosh violations, Lipinski violations, etc. Ligands/natural compounds were selected based on adherence to soft or classical Lipinski's rule of five. The selected ligands that did not incur more than 2 violations of Lipinski's rule were further used in molecular docking experiments with the target protein.
2.5. Target prediction
Molecular target studies are important to find the macromolecular targets of bioactive small molecules. This is useful to understand the molecular mechanisms underlying a given phenotype or bioactivity, rationalize possible side-effects, and predict offtargets. In this direction, SwissTargetPredictiontool (https://www.swisstargetprediction.ch) was used[15]. Canonical SMILE for amentoflavone and guggulsterone was entered and was analyzed.
2.6. MD simulations
The four representative docking complexes of ligands with Mincluding amentoflavone, guggulsterone, puerarin, and piperine were used for further refinement using MD simulations analysis.MD simulation studies were carried out to find out the stability and flexibility of the natural compounds-Mcomplexes at 50 ns.The method for the MD simulations of natural compounds-Mcomplexes was the same as recent studies[16,17]. All simulations of representative natural compounds-Mcomplexes were conducted with the utilization of GROMOS96 43a1 force field available in GROMACS 5.1.4 suite[18]. Topology files for ligand molecules were created by using the PRODRG server[19]. The prepared protein complexes were solvated in a cubic box of edge length 10 nm along with simple point charge water molecules. To maintain the system neutrality, adequate numbers of ions were added. To remove the clashes between atoms, system energy minimization calculations were applied with the convergence criterion of 1 000 kJ/mol/nm. Long-range interaction electrostatics[20]was handled by using particle mesh Ewald. For both van der Waals and Coulombic interactions, a cut-off radius of 9 Å was utilized. Equilibration was completed in two different phases. The solvent and ion molecules were kept unrestrained in the first stage, while in the second stage the restraint weight from the protein and protein-ligand complexes was gradually declined in the NPT (constant number of atoms N,pressure P, and temperature T) ensemble. LINear Constraint Solver constraints were applied to all bonds involving hydrogen atoms[21].The temperature and pressure of the system were kept at 300 K and 1 atm, respectively by using Berendsen's temperature and Parrinello-Rahman pressure coupling, respectively[22]. The production simulation was initiated from the velocity and coordinates obtained after the last step of the equilibration step. All the systems were simulated for 200 ns and snapshots were taken at every 2 ps interval.The conformational stability and flexibility of the complexes were analyzed by various parameters such as root mean square deviation(RMSD), root mean square fluctuation (RMSF), solvent accessible surface area (SASA), radius of gyration (Rg), and binding affinity of phytomolecule complexes by using MMPBSA and hydrogen bond formation ability. The RMSD is a commonly used similarity tool to measure the conformational perturbation during the simulation of macromolecule structures.
2.7. Free energy calculations by MM/PBSA
The calculations of the binding energy of the M-ligand complexes were calculated by using MM/PBSA method. While the polar part of the solvation energy was calculated by using the linear relation to the solvent accessible surface area. The g_mmpbsa module available in GROMACS was applied for the determination of different components of the binding free energy of complexes[23].Only data of the last 10 ns were utilized for the MM-PBSA analysis considering the convergence issue associated with the calculations.In the present study, entropy calculations were not performed as they may change the numerical values of the binding free energy reported for the molecules. In the MM-PBSA calculation, the binding free energy between Mand a ligand was calculated using the following equations:

Where ∆G: Free energy of molecular mechanics Poisson-Boltzmann surface area; G: the total free energy of the proteinligand complex; Gand Gare total free energies of the isolated protein and ligand in solvent; Gis the free energy of system x that being the ligand, the protein, or the complex; T is the temperature; Sis the entropy; and angle brackets represent an ensemble average. Eis the potential energy in vacuum as defined by the molecular mechanics (MM) model, which is composed of the bonded energy terms (E) and nonbonded Coulombic (E) and Lennard-Jones (E) terms; ∆G= Free energy of solvation is the combination of Gand G.
2.8. Toxicity analysis
The prediction of compound toxicities is an important parameter for drug design process. In order to check toxicity of these selected natural compounds, ProTox-Ⅱ web server was used[24]. ProTox-Ⅱis a kind of virtual lab that integrates several parameters like molecular similarity, fragment propensities, and most frequent features. It predicts various toxicity endpoints and incorporates a total of 33 models for the prediction of various toxicity aspects of small molecules.
2.9. Comparison with similar FDA approved drug compound by SwissSimilarity
The compounds which had suitable parameters including ADME& toxicity and the best binding energy were compared with FDA-approved drugs using SwissSimilarity tool (http://www.swisssimilarity.ch)[25].
2.10. Computational facility details
The MD simulations and corresponding energy calculations were carried out on HP Gen7 server with 48 Core AMD processors and 32GB of RAM.
3. Results
3.1. Determination of active sites
The structure and amino acids were found in the active site pockets of 6LU7.
3.2. ADME
The drug scanning results showed that most of the tested compounds were accepted based on Lipinski's rule of five. These compounds were selected for docking to find their binding affinity with COVID19 main protease M. The list of compounds with suitable ADME properties is given in Supplementary Table 2.

Table 2. Binding free energy calculation of four stable complexes during simulation (kJ/mol).
3.3. Target prediction
The target prediction analysis was performed for two best compounds, amentoflavone and guggulsterone on the web page.For amentoflavone, it predicted 20% of family AG proteincoupled receptor, 13.3% of kinase, 13.3% of enzymes, 13.3% of unclassified protein, 6.7% of phosphatase, 6.7% of protease,6.7% of oxidoreductase, 6.7% of primary active transporter,6.7% of secreted protein, and 6.7% of ligand-gated ion channel.While for guggulsterone, it predicted 40% of nuclear receptors,13.3% of cytochrome P, 13.3% of secreted protein, 13.3% of oxidoreductase, 6.7% of membrane receptors, 6.7% of fatty acidbinding protein family, and 6.7% of enzymes. The details including target, common name, UniProtID, ChEMBLID, target class,probability, and known actives in 2D/3D are given in Supplementary Table 3 & 4. The possible sites of the target which the compound may bind to were mostly the targets predicted by the software. The probability scores for amentoflavone and guggulsterone were from 1.000 to 0.087 & 1.000 to 0.102, respectively.
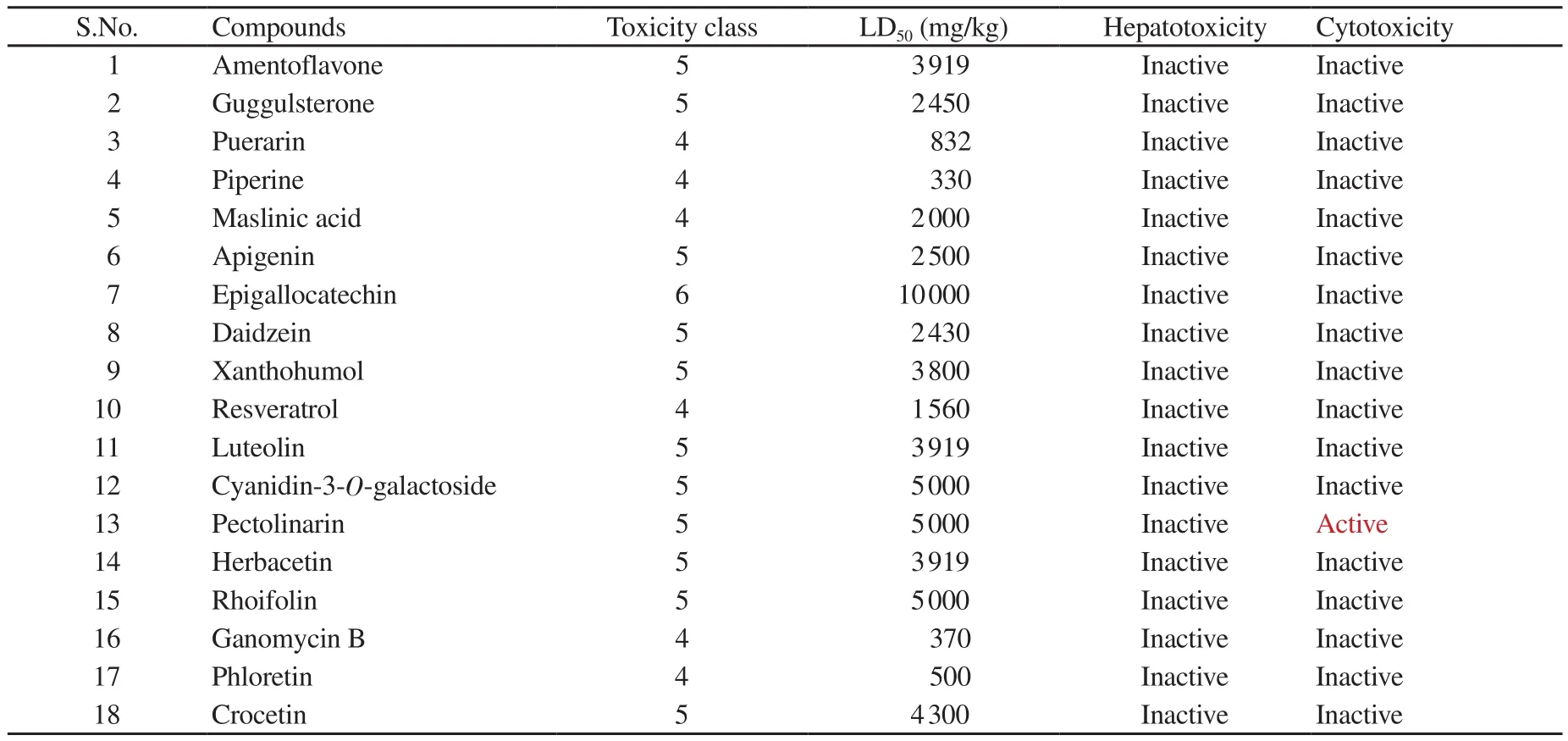
Table 3. Toxicity predictions for selected natural compounds.
3.4. Molecular docking
A total of 18 selected natural compounds were docked with COVID-19 main protease Mwith ritonavir and lopinavir as standard controls. The results indicated a good binding affinity of ritonavir and lopinavir to the COVID-19 main protease M.The docking results are given in Table 1 and docking analysis visualization is shown in Figure 1. Molecular docking results between COVID-19 main protease M(PDB-6LU7) and selected natural compounds and schematic representation of molecular docking between Mand top four natural compounds are given in Supplementary Figure 2 & 3.

Figure 2. (A) Root mean square deviation (RMSD) backbone; (B) RMSD ligand; (C) root mean square fluctuation (RMSF); (D) radius of gyration for all four complexes over the 50 ns simulations.

Figure 3. (A) Solvent accessible surface area (SASA); (B) hydrogen bond numbers; (C) hydrogen bond distribution for all four complexes during MD simulations for 50 ns.
3.5. MD simulations
The time-dependent RMSD was calculated from the initial stage of the simulation on 50 ns. The RMSD of the backbone of these 4 complexes was 0.2 to 0.5 nm (Figure 2A), which stabilized at the 35 ns whereas the RMSD of ligands ranged from 0.3 to 1.0 nm (Figure 2B). During MD simulations, RMSF defines the residual flexibility from the average position. The RMSF of the protein ranged from 0.1 to 0.5 nm of all systems (Figure 2C). Some amino acids showed a high-intensity peak, which may represent a loop region.In MD simulations, Rg is a well-known method to determine the compactness of protein. The compactness of a protein is induced by the movement of a ligand. During simulation analysis on 50 ns, the lower flexibility of the Rg is associated with the structural stability of the protein. The Rg values of all phytochemical complexes were 2.20 to 2.05 nm (Figure 2D). The Rg values of all four phytochemical complexes supported their consensus architecture as well as size.The predicted SASA values were found to be associated with the exposures of hydrophobic residues during simulation analysis.SASA plays a principal role in the van der interaction. The SASA values of all systems were 125-150 nm(Figure 3A).
In a complex protein and ligand, hydrogen bonding plays a critical role in determining the strength of interaction. During the simulation time, several hydrogen bonds formed between the donor and the acceptor group (Figure 3C). Two hydrogen bonds were consistently formed during the time of simulation (Figure 3B). Overall observations indicated that all four complexes were stable during simulation.
3.6. Binding free energy calculation
The free binding energy results showed that as compared with other molecules, amentoflavone had maximum binding energy followed by guggulsterone (Table 2). In each molecule, the polar salvation and SASA energy showed moderate effects on binding energy component.
3.7. Toxicity analysis
In-silico toxicities of selected natural compounds were predicted by using ProTox-Ⅱ. As shown in Table 3, none of the selected natural compounds showed potential hepatotoxicity or cytotoxicity except pectolinarin.
We further checked the similarity of two top natural compounds amentoflavone and guggulsterone with the FDA approved drugs using SwissSimilarity check. For amentoflavone, we did not find any reported similar FDA approved drug. Whereas for guggulsterone,we found 117 FDA proved drugs. The details including drug ID,drug name, similarity score, and molecule structure are given in Supplementary Figure 4 & 5. The structural similarity of other FDA approved drugs with guggulsterone was predicted using SwissSimilarity and the probability score obtained was 0.995 to 0.009.
4. Discussion
COVID-19 pandemic is declared as a public health emergency and is lack of specific treatment[3]. There is a dire need for effective drugs against the novel coronavirus COVID-19. Since the virus is new, information about its molecular mechanism is very limited.The only reference is from the old SARS virus (SARS-CoV-1)that emerged in 2003. Fortunately, in a recent study, Jin et al. have revealed the crystal structure of SARS-CoV-2/COVID-19 main protease (M/3CL protease, PDBID-6LU7)[8]. This protease is considered as an important target as it is essential for virus functionality, replication, and entry competence. The main protease Mhas been investigated as a potential target to inhibit previous coronavirus infections like SARS and MERS[26]. This study aimed to screen the natural compounds based on their pharmacokinetic properties, drug-likeness, and ability to specifically bind to the active sites of SARS-CoV-2 main protease. Target prediction suggests that the small compound may have high target attraction towards the specific binding site. To further investigate the molecular docking results, the top four natural compound complexes namely,amentoflavone, guggulsterone, puerarin, and piperine were subjected to 50 ns MD simulations. RMSD of the Cα atoms is related to the stability of the complexes. RMSD value may indicate that the binding of ligands does not lead to the conformation perturbation during the simulation. RMSD of the protein backbone in all systems was small and comparable. The presence of low-intensity peak suggested that binding of the phytomolecules does not affect the stability of the structural region of the enzyme. SASA conformations showed that the binding of ligand molecules does not affect the overall folding of the protein. Lopinavir and ritonavir are wellknown protease inhibitors of human immunodeficiency virus[27].Both drugs were also recommended as repurposed drugs in the treatment of SARS and MERS[5]. Therefore, in this study, we took these drugs as standard reference drugs for comparison. In our insilico prediction experiment, none of the selected compounds showed hepatotoxicity and cytotoxicity except pectolinarin. The compounds with potential to inhibit the viral protease based on the binding energy include amentoflavone, guggulsterone, puerarin, piperine,maslinic acid, apigenin, epigallocatechin, daidzein, xanthohumol,resveratrol, luteolin, cyanidin-3-O-galactoside, pectolinarin,herbacetin, rhoifolin, ganomycin B, phloretin, and crocetin. Among these compounds, amentoflavone and guggulsterone were the top two with the best binding energy and most satisfactory parameters.Guggulsterone from a Indian traditional ayurvedic medical plant Commiphora mukul was found to be the most suitable based on comprehensive pharmacokinetic parameters, drug-likeness, and docking analysis. Therefore, we propose guggulsterone as a potential inhibitor of COVID-19 main protease M. Further in-vitro and invivo studies are still needed to validate these bioinformatics based findings.
Conflict of interest statement
We declare that there is no conflict of interest.
Authors' contributions
VT conceived the idea, wrote the discussion part,and supervised the entire study. AM and YP contributed to the research tool and wrote the manuscript. AK & SKM did the critical analysis.
 Asian Pacific Journal of Tropical Biomedicine2021年4期
Asian Pacific Journal of Tropical Biomedicine2021年4期
- Asian Pacific Journal of Tropical Biomedicine的其它文章
- Cannabinoid CB2 receptors and spinal microglia are implicated in tingenonemediated antinociception in mice
- Morin attenuates L-arginine induced acute pancreatitis in rats by downregulating myeloperoxidase and lipid peroxidation
- Lentinula edodes extract inhibits matrix metalloproteinase expression and increases typeⅠprocollagen expression via the p38 MAPK/c-Fos signaling pathway in ultraviolet A and B-irradiated HaCaT keratinocytes
- Reactive oxygen species and senescence modulatory effects of rice bran extract on 4T1 and NIH-3T3 cells co-treatment with doxorubicin