
遗传改造微生物制造食品和饲料的监管要求及欧盟授权案例分析
2021-03-18 07:52魏笑莲钱智玲陈巧巧于洪巍
合成生物学 2021年1期
魏笑莲,钱智玲,陈巧巧,于洪巍
(1 浙江新和成股份有限公司,浙江 新昌 312500;2 浙江大学化学工程与生物工程学院生物工程研究所,浙江 杭州 310027)
遗传改造技术在食物供给中正被日益广泛地使用。遗传改造技术在带来便利和巨大利益的同时,也给人类社会带来了种种风险和挑战。遗传改造的生物可能会对人类和动物产生潜在的毒性、过敏性或其他有害影响,也可能产生“基因漂移”和“基因污染”现象,对生态环境造成影响[1]。因此,许多国家都建立了相应的法律法规来监管遗传改造生物及其产品。不同国家历史文化背景、技术水平和公众对遗传改造食品的认知程度不同,对遗传改造食品的监管也存在着截然不同的做法。
本文主要讨论对象为遗传改造微生物(genetically modified microorganisms,GMMs)发酵生产的食品、饲料监管要求。对于遗传改造微生物发酵生产的食品、饲料产品,若最终产品中能检测出来自于遗传改造微生物的组分[通常指GMMs 及重组DNA(rDNA)],则在欧盟、美国被归属为转基因食品(genetically modified food,GMF),需要符合转基因食品相关法律法规的监管要求。本文首先介绍欧盟、美国和中国对转基因食品的定义和监管要求,再以监管最为严格的欧盟为例,深入剖析遗传改造微生物发酵生产的食品、饲料在欧盟的监管、授权要求。
1 转基因食品定义
不同国家对转基因食品的理解和定义不同。
关于欧盟,根据欧盟第1829/2003 号关于转基因食品和饲料的法规,包含转基因生物、由转基因生物构成或由转基因生物加工而成的食品和饲料称为转基因食品和饲料。值得注意的是,该法规指出由转基因生物加工而成(food and feed produced ‘from’ a GMO)与由转基因生物发酵生产(food and feed produced‘with’a GMO)的区别。认定标准是食品或饲料中是否存在转基因生物的组分(如转基因微生物的菌体及rDNA)或加工而成的成分(如转基因玉米加工而成的玉米制品)。例如,如果产品由转基因微生物发酵生产,且产品中没有检测到转基因微生物以及rDNA,则产品不符合欧盟关于转基因食品和饲料的定义,不受欧盟1829/2003 号法规的监管,也不需要满足转基因食品饲料相关的标识要求。反之,若产品中检测出了转基因微生物及rDNA,则产品符合欧盟关于转基因食品和饲料的定义,需要符合欧盟1829/2003 号法规的监管要求。同时,该法规考虑到一些生产者并不使用转基因食品和饲料,但由于在种子生产、栽培、收获、运输或加工过程中的偶然或技术上不可避免地存在转基因物质,这种转基因物质可能在非转基因的传统食品和饲料中以微量存在。因此,该法规也规定如果是偶然或者技术上不可避免的原因,转基因组分或加工而成的成分含量在单独的食品成分或由单个成分构成的食品中不超过0.9%的阈值,则不需要符合欧盟1829/2003号法规的监管要求。
在美国,为了让消费者更容易接受转基因食品,将转基因食品称作生物工程食品(bioengineered foods)。美国2018年12月20日宣布了《国家生物工程食品信息披露标准》(National Bioengineered Food Disclosure Standard)。该标准规定了生物工程食品的标签要求,并将生物工程食品定义为含有可检测的遗传改造物质的食品,且这些遗传改造物质通过某些实验室技术进行了修饰,不能通过常规育种得到或在自然界中发现。该标准要求,到2022年,食品制造商、进口商和某些零售商必须要为生物工程食品或含有生物工程成分的食品贴上标签。值得注意的是,美国法规将膳食补充剂包括在该标准涵盖的食品范围。但是如果食品是经过高度精制,并且不含可检测的遗传改造物质,则不属于生物工程食品。与欧盟类似,美国法规规定如果传统非生物工程食品中,无意或技术上不可避免地混入了遗传改造物质,只要遗传改造物质的含量不超过5%,则可以豁免于该法规监管,但5%的范围相比欧盟的0.9%要宽泛许多。例如,如果食品制造商采购非生物工程玉米,而该非生物工程玉米的生物工程物质的含量在5%内,则为无意或技术上无法避免的,则无需披露。但是,如果食品制造商打算使用由生物工程食品(例如玉米)生产高度精制的成分,但该成分精制后仍可检测到遗传改造物质,无论该成分的含量如何,仍需要披露该成分。
在中国,现行有效的法律法规并未明确定义转基因食品及其范围。我们仅能通过已经被废止的几个规定找到一些线索。根据2002 年原卫生部发布的《转基因食品卫生管理办法》,“转基因食品系指利用基因工程技术改变基因组构成的动物、植物和微生物生产的食品和食品添加剂,包括转基因动植物、微生物产品;转基因动植物、微生物直接加工品;以转基因动植物、微生物或者其直接加工品为原料生产的食品和食品添加剂。然而在《转基因食品卫生管理办法》被废止后,更新的法规均未对转基因食品重新进行定义。目前我国转基因食品立法和制度化建设滞后于欧美等国家和地区,对转基因食品的定义、管理缺乏明确的制度化规定。
综上,我们可以概括地将转基因食品理解为包含可检测到遗传改造物质的食品。
2 各国家和地区对转基因食品的监管力度
因各国的历史文化背景、技术水平和公众对转基因食品的认知程度不同,各国采取的监管模式也有所不同。主要分为欧盟的严格监管模式和美国的宽松监管模式,其他的国家基本介于两者之间。
欧盟基于“谨慎预防原则”,采取严格的监管模式。欧盟制定了一系列完善的法律法规和配套指南来对转基因食品进行严格监管。只要涉及到转基因,无论产品之前在其他国家是否被批准上市,无论产品中是否能检测到转基因成分,在产品上市之前都需要经过欧洲食品安全局的评估和审核。只有得到欧洲食品安全局和欧盟委员会的批准后才允许上市。这种基于“谨慎预防原则”的管理模式,将转基因食品可能带来的危害降低,考虑到科学的局限性和不确定性,认识到转基因食品对于人类健康、生态环境的威胁,而受到不少学者的推崇[2-3]。
美国基于“实质等同”原则,认为传统食品与转基因食品不存在本质区别,对转基因食品采取宽松的监管模式。美国食品药物监督管理局(FDA)、美国环境保护署(EPA)和美国农业部(USDA)共同负责转基因食品的监管。美国食品药物监督管理局(FDA)监管大多数人类和动物食品,包括转基因食品,确保转基因食品或含有转基因成分的食品符合与所有其他食品相同的严格安全标准。也就说美国FDA 没有制定专门的法规来监管转基因食品,而是将转基因食品纳入现有的法律框架,转基因食品和普通食品一样遵循相同的安全标准要求。随着《国家生物工程食品信息披露标准》(National Bioengineered Food Disclosure Standard)的颁布,美国对转基因食品的标识监管在逐步加强。然而FDA 目前的政策中仍然强调转基因食品的加工方法并不是监管重点,只要特性和化学组成与传统食品一致,就不需要对转基因食品进行入市前的审查。
我国转基因相关的法律法规所规范对象主要是农业转基因生物。根据《农业转基因生物安全管理条例》,农业转基因生物是指“利用基因工程技术改变基因组构成,用于农业生产或者农产品加工的动植物、微生物及其产品,主要包括:①转基因动植物(含种子、种畜禽、水产苗种)和微生物;②转基因动植物、微生物产品;③转基因农产品的直接加工品;④含有转基因动植物、微生物或者其产品成分的种子、种畜禽、水产苗种、农药、兽药、肥料和添加剂等产品。”从上述定义可以看出,农业转基因生物实际上是一个非常宽泛的概念,所涵盖的产品范围较为宽泛,且似乎没有将作为深加工产品“食品”大类(不含上述定义中明确列入的农产品和直接加工品)明确纳入农业转基因生物。从上述定义中,农业转基因生物,首先需要利用基因工程技术改变基因构成,其目的是用于农业生产或者农产品加工。而市场上销售的预包装食品等,以及《食品安全法》下的食品添加剂应不包含在农业转基因生物的定义中。我国目前并未明确定义转基因食品,没有单独的转基因食品法。并且转基因食品安全监管存在立法内容不全、立法层次较低、监管制度缺陷等问题[4]。因此国内诸多学者建议通过进一步健全现有法律制度、加强立法的系统性[5]、增进监管制度的民主性[6],来完善我国转基因食品安全监管。
综上,欧盟对转基因食品的监管最为严格,鉴于欧盟市场的重要性,下文将从欧盟的食品安全风险分析框架着手,概述欧盟遗传改造微生物发酵食品及饲料产品的授权过程。
3 欧盟转基因食品安全监管主体
3.1 欧盟理事会
欧盟理事会(Council of European Union)由欧盟各成员国部长组成,因此又称“部长理事会”(the Council of Ministers),是欧盟的重要决策机构。欧盟理事会负责日常决策并拥有欧盟立法权。在转基因食品监管中,欧盟理事会承担制定相关法规,总体规划和协调的作用,如授权欧盟委员会执行相关法律法规,邀请成员国充分参与转基因食品的全程监管等。
3.2 欧洲议会
欧洲议会(European Parliament)是欧盟三大机构(欧盟理事会、欧盟委员会、欧洲议会)之一,为欧盟的参与立法、监督、预算和咨询机构。欧洲议会共有20 个专门委员会,其中环境、公共卫生和食品安全委员会、农业委员会、内部市场和消费者保护委员会等与转基因食品安全监管相关。
3.3 欧盟委员会
欧盟委员会(European Commission),是欧盟的常设执行机构,也是欧盟唯一有权起草法令的机构。欧盟委员会监督各欧盟成员国对欧盟法律的履行,作为欧盟执行机构,其负责欧盟各项法律文件(指令、条例、决定)的具体贯彻执行。在转基因食品监管中,欧盟委员会负责相关法律法规的起草及贯彻执行。
3.4 欧洲食品安全局(European Food Safety Authority,EFSA)
EFSA 是欧洲食品安全系统的主要单位,EFSA 于2002 年成立,EFSA 成立的主要目的是提供独立完整的科学意见,让欧盟决策单位面对食物链直接与间接相关问题及潜在风险能做出适当的决定,以提供欧洲公民安全高品质的食物。在转基因食品监管中,EFSA 评估转基因生物对欧洲人类和动物健康以及环境的潜在风险。EFSA 的作用是向欧洲风险管理者(例如,欧盟委员会和欧盟成员国)就转基因生物的安全性提供科学建议。EFSA 采用欧盟监管框架中规定的严格标准评估转基因生物的安全性,通过评估后才有可能授权将它们用作食品或饲料或在欧盟进行种植。在风险管理者做出市场授权决定之前,EFSA 会评估转基因产品的安全性。该评估包括对转基因生物对人类健康,动物健康和环境的潜在影响的评估。EFSA 的评估基于申请人提交的科学档案和任何其他相关科学信息。风险管理者授权转基因生物后,通常会获得欧盟市场10 年的许可。10 年后,必须先由EFSA 重新评估,然后再做出任何重新授权决定。同时,为帮助申请者理解法规要求顺利通过评估,EFSA 为转基因生物及其衍生食品和饲料的风险评估准备了指导文件,这些文件详细说明了如何编写转基因产品申请卷宗以及卷宗中需要包括的科学数据。所有指导文件均可在EFSA 网站上公开获得。值得注意的是,EFSA会经常更新其指导文件,申请者在申请前务必确保使用最新版本。表1 列出了欧洲食品安全局关于转基因食品授权的主要指导文件,截止到目前都是现行有效版本。本文侧重于分析遗传改造微生物发酵生产的食品和饲料欧盟授权要求,尤其是对遗传改造微生物自身的要求,故而下文将重点分析EFSA 于2011 年6 月颁布的食品和饲料用遗传改造微生物及其产品风险评估指南以及2018 年2 月颁布的用作饲料添加剂或生产生物的微生物特性描述指南,以帮助申请者更好地理解EFSA 对发酵菌种的要求。
4 欧盟遗传改造微生物发酵食品、饲料授权要求
4.1 食品和饲料用遗传改造微生物及其产品风险评估指南
GMMs参与多种食品和饲料的生产。这些产品在欧盟的上市批准由不同的法律法规监管,需要满足不同的风险评估要求。欧洲食品安全局2011年颁布食品和饲料用遗传改造微生物及其产品风险评估指南,指南根据产品的性质和EFSA转基因生物小组评估所需的科学信息水平,将产品分为4类(表2),每类产品需要提供不同的数据(表3)。
除了提供表3中要求的资料外,属于第一类和第二类饲料添加剂的氨基酸和酶以及属于第四类的微生物饲料添加剂根据欧盟委员会第429/2008号法规(关于欧盟第1831/2003 号的实施细则)进行评估,该条例涉及饲料添加剂的评估和授权。属于第三类的作为饲料用途的生物质根据欧盟第1829/2003号法规评估。属于第二类的食品用酶根据欧盟第1332/2008号法规和第1331/2008号法规评估。属于第一类或第二类的食品添加剂应符合欧盟第1333/2008号法规和第1331/2008号法规的要求。属于第一类或第二类的食用香料应符合欧盟第1334/2008号法规和第1331/2008号法规的要求。
4.2 食品和饲料用遗传改造微生物发酵产品特性描述要求
如前所述,遗传改造微生物发酵的产品所属类别不同,授权需要符合的法规要求、需要提供的资料详实程度也不同。如果产品中含有转基因成分,则需要遵从欧盟第1829/2003 号法规要求;如果产品中不含有转基因成分,则根据产品的类别遵从不同法律法规的要求。EFSA 于2018 年2 月颁布了用作饲料添加剂或用作生产生物的微生物特性描述指南,相比于EFSA 2011年颁布的食品和饲料用遗传改造微生物及其产品风险评估指南,2018年的指南结合最近的科学发展和评估经验更加详细地描述了用于饲料添加剂生产的微生物特征要求。虽然该指南的目的是帮助申请者准备和提交饲料添加剂的授权申请,但考虑到食品添加剂等产品
也是由EFSA 监管,我们在准备食品类产品的批准和授权时也可以参照该指南的要求进行准备。结合2011年的和2018年的指南,可以看出EFSA对于遗传改造微生物的风险评估过程是基于循序渐进和基于个案原则进行的,对于最终产品中不含转基因成分的遗传改造微生物发酵产品(第一类)的菌种特征描述要求可以简要归纳如下。

表1 欧洲食品安全局关于转基因食品授权的主要指导文件Tab.1 Main guidance documents of European Food Safety Authority on authorization of genetically modified foods

表2 EFSA食品和饲料用遗传改造微生物及其产品风险评估指南中对产品的分类Tab.2 Classification of products in EFSA guidance on the risk assessment of genetically modified microorganisms and their products intended for food and feed use

表3 申请将转基因产品及其衍生食品和饲料产品投放欧盟市场所需的微生物信息Tab.3 Microbial information required for the placing on the market of GMMs and their derived food and feed products
4.2.1 受体或亲本微生物的特征
对于第一类产品,受体或亲本微生物的特征需要提供的资料包括亲本微生物的学名、分类学和其他名称、表型特征及亲本菌株基因改造的历史。需要提供以下分类信息:①属;②种;③亚种(如适用);④株;⑤保藏号;⑥通用名;⑦商品名。表型特征可包括形态、生长要求、生长速率、温度及pH 范围和最佳值、孢子形成能力、有氧和/或无氧代谢、抗性特性等。相关遗传标记指营养缺陷型突变和编码抗性基因的特征等。在进行亲本微生物特征描述时可以参照2018 年的指南的相关规定包括如下。
(1)WGS(whole genome sequencing,全基因组测序)鉴定 WGS 法是评估微生物特性的首选方法,这对于细菌和酵母是强制性的,对于丝状真菌也推荐使用WGS分析。WGS应优先用于鉴定微生物和记录基因改造。WGS 可以提供菌株的详细的分类学鉴定信息,还可以借助WGS 分析抗性基因和毒力/产毒性因子的相关基因。因此,它是了解遗传改造微生物风险评估的一个基本要素。
(2)QPS(qualified presumption of safety,合格安全假定)方法评估 QPS 的评估是基于广泛的文献搜索,以发现相关的人类、动物和环境的可能安全问题。符合QPS 方法的菌株被认为对目标物种、消费者和环境是安全的,无需具体研究。如果受体菌株符合QPS 的条件,并且如果基因改造没有问题,那么QPS状态也适用于转基因生物。
(3)抗生素敏感性 对于微生物抗性的风险评估重点在于区分固有抗性和获得性抗性。使用组合方法进行表型测试,基于最低抑菌浓度MIC测定,与区分敏感菌株和抗性菌株的规定临界值进行比较,以及WGS分析。
(4)抗菌活性 添加剂不应产生与人类和动物使用相关的抗菌剂。应通过对照已知对一系列抗生素敏感的参考菌株测试培养上清液来证明不存在抗微生物活性。如果在一个或多个物种中有阳性结果,应鉴定抑制物质。
(5)菌种毒力和致病性分析 在指南中,对产毒性和致病性的评估基于3种不同的证据收集方法,这些方法应在证据权重方法中使用。第一是基于文献检索菌株或其亲缘相近的菌种的使用历史。第二个是基于WGS 生物信息学分析已知毒力因子或有毒化合物的存在。第三是基于模型生物进行微生物毒性测试。
4.2.2 插入序列(供体生物体)的特征
插入序列的来源和功能对于确定基因产物对人类、动物、植物健康和环境的潜在毒性、毒力或过敏性很重要。插入GMM 中的序列可以源自特定的生物体或设计序列。当插入的DNA 是来自不同来源的序列的组合时,应提供每个序列的相关信息。来自特定供体的DNA 序列的描述应包括插入元素的核苷酸序列、结构和功能、编码蛋白的名称等。设计序列是在自然界中未知的序列,例如密码子优化的基因。设计序列应提供设计的原理和策略,序列和功能元件的物理图谱,编码蛋白的衍生氨基酸序列和功能,与数据库中的序列相似性等。
4.2.3 遗传改造的描述
遗传改造应详细描述改造方案,当使用质粒作为载体时,对载体的特征、来源及风险评估也应详细描述。遗传修饰的过程描述,包括基因序列插入、删除、替换或修饰的方法等。
4.2.4 遗传改造微生物相关的信息
经过改造的GMM 需存放在欧盟认可的菌种保藏机构,申请授权时需提供保藏编号。另外,还应提供以下与GMM 相关的信息:①遗传性状或表型特征的描述,尤其是基因修饰所引起的可能被表达或不再被表达的性状或特征;②GMM 中载体和/或供体DNA 的结构和数量,当载体或供体DNA 中存在抗性标记时,这一点尤为重要;③识别和检测插入序列和载体的技术。
4.2.5 与产品相关的信息
(1)与生产过程相关的信息 应该详细描述遗传改造微生物生产过程相关的信息如发酵、培养的流程并推荐显示关键阶段的流程图。
(2)与产品制备过程相关的信息 应提供与产品制备相关的信息和用于去除GMM 细胞和重组基因的详细技术信息。应证明去除方法的可靠性和有效性,确保产品中不含转基因生产菌株。此外应提供产品中重组DNA 可能存在的信息。如果发现了与全长编码序列相对应的重组DNA,必须评估基因转移的可能性。
4.3 遗传改造微生物发酵食品、饲料授权流程
遗传改造微生物发酵产品所属类别不同,监管法规不同,授权的过程也略有差异。但产品的授权都必须先通过EFSA 的安全性评估后才有可能合规在欧盟上市销售。以饲料添加剂为例,欧盟法规EC 1831/2003 中描述了授权的申请程序。申请人必须:①向欧盟委员会提交申请;②直接向EFSA 提交技术档案;③向欧盟参考实验室提交三份饲料添加剂参考样品。欧盟委员会网站提供了饲料添加剂授权程序的概述。EFSA 进行风险评估,EFSA 在收到卷宗后6 个月内出具意见,并会针对卷宗提出问题要求补充资料。而欧盟委员会在收到EFSA 观点后的三个月起草法规文件,建议成员国批准或拒绝该授权。对于任何类型的饲料添加剂申请,申请人应参考关于动物营养添加剂的第1831/2003 号法规及第429/2008 号法规的实施细则,以及EFSA 的相关行政指导和技术指导文件来准备和提交档案。EFSA 关于饲料添加剂应用的行政和技术指导文件定期更新,因此提醒申请人在申请时务必确保使用这些文件的最新版本。值得注意的是,饲料添加剂类的授权有效期为10年,最迟应在授权到期日前一年向欧盟委员会提交更新申请。
5 欧盟遗传改造微生物发酵产品授权和拒绝实例分析
遗传改造微生物参与多种食品和饲料的生产。在食品和饲料工业中,遗传改造微生物可用于生产酶制剂、氨基酸、有机酸、维生素等。大肠杆菌等原核表达系统具有遗传背景研究清楚、操作简单、生长繁殖快、成本低、产量高、表达产物纯化过程简单、产物稳定性好、不易污染以及应用范围广等优点,是最早进行研究的外源基因表达系统,也得到了非常广泛的应用,取得了巨大的科研价值和经济效益。其在生物、食品、工业以及医学领域发挥着举足轻重的作用[15]。然而许多大肠杆菌菌株是人类和其他动物的病原体,可导致泌尿和肠道疾病,也常是败血症和许多其他全身感染的原因。在欧盟,EFSA 决定将这一物种排除在未来的QPS评估之外[16],安全性评估必须在菌株水平上进行[17]。在2020 年EFSA 发布的最新QPS清单中,因为分类地位不明确或具有潜在的有害特征,EFSA还将filamentous fungi、bacteriophages、Streptomycetes、Oomycetes、Enterococcus faecium等排除在QPS 评估之外[18]。然而这并不意味着大肠杆菌不允许用作生产菌株发酵生产食品和饲料产品,只要证明菌种的安全性,仍然有可能被欧盟授权。表4 汇总了2019—2020 年EFSA 收到的部分大肠杆菌发酵菌株申请情况,这些申请均在评估中,尚未获得EFSA 认可及欧盟委员会的正式授权和批准。
此外,我们也查询了2019—2020 年由欧盟委员会正式授权的由大肠杆菌生产的产品,部分结果汇总见表5。从汇总结果表中我们可以看出,所授权的大肠杆菌菌株都是基于大肠杆菌K-12 菌种改造的,大肠杆菌K-12 具有很好的特性,其安全性(非致病性)已被广泛记录,如果使用大肠杆菌K-12 作为亲本菌株进行改造,会相对容易通过EFSA 的评估。值得注意的是,遗传改造的大肠杆菌CGMCC 11473 生产饲料添加剂L-苏氨酸的授权实例。申请者在首次申请时,EFSA 表示申请者所提供的信息不足以描述遗传改造的特征。在EFSA 推荐的10 种临床相关抗菌药物敏感性检测中,申请者只检测了其中的5 种,并且卡那霉素的最小抑菌浓度MIC 为256 mg/L,超过了临界值。然而申请者没有提供关于卡那霉素抗性的遗传基础信息,没有在生产菌株的基因组中搜索赋予卡那霉素或其他抗性的基因。因此,生产菌株中存在抗性基因的可能性仍然存在。并且申请者没有提供亲本菌株、受体菌株和遗传改造过程的信息。生产菌株的细胞及其重组DNA 在产品中的存在仍然存在不确定性。因此,EFSA 无法就由大肠杆菌CGMCC 11473 发酵生产的L-苏氨酸产品对目标物种、消费者和环境安全性得出结论[19]。再次申请时,申请人补充提供了关于生产菌株的身份、遗传修饰、抗生素敏感性以及最终产品中生产菌株的细胞和重组DNA 缺失的额外数据。EFSA 认为受体生物大肠杆菌K-12 是安全的。遗传修饰增加了L-苏氨酸的生物合成能力。生产菌株中没有保留编码抗性的基因。受体菌株是安全的,遗传改造不会引起问题。此外,在最终产品中没有检测到生产菌株的活细胞或DNA。根据这些新信息,EFSA 专家组得出结论,大肠杆菌CGMCC 11473 生产的L-苏氨酸对所有动物物种、消费者和环境都是安全的[20]。从这些案例中我们了解到,申请者必须确保提供的信息符合相关法规和指南的要求,切不可存在侥幸心理提供不完整的评估卷宗,以期通过EFSA 审核,这样只会延长评估和授权的周期。另外,如果基因修饰过程没有引入抗性基因,并且最终产品中没有生产菌株的活细胞及其DNA,EFSA 评估通过的可能性也会大大提高。

表4 2019—2020年EFSA 收到的部分大肠杆菌发酵产品授权申请清单Tab.4 List of application for some products produced by fermentation of E.coli received by EFSA from 2019 to 2020
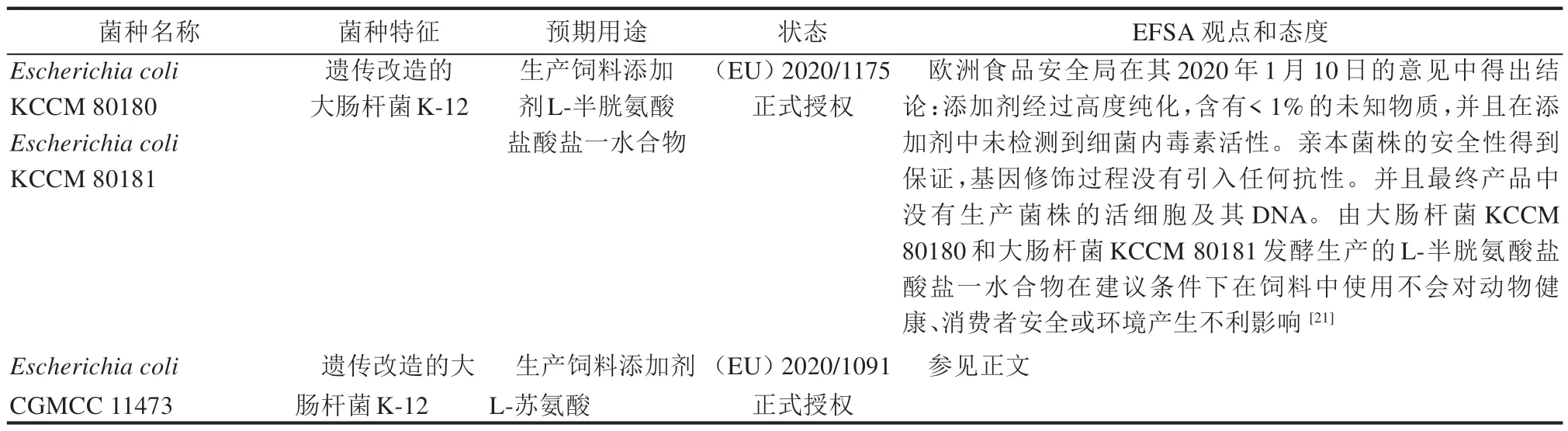
表5 2019—2020年欧盟委员会批准的部分由大肠杆菌生产的产品清单Tab.5 List of authorizations by EU from 2019 to 2020 for some products produced by fermentation of E.coli
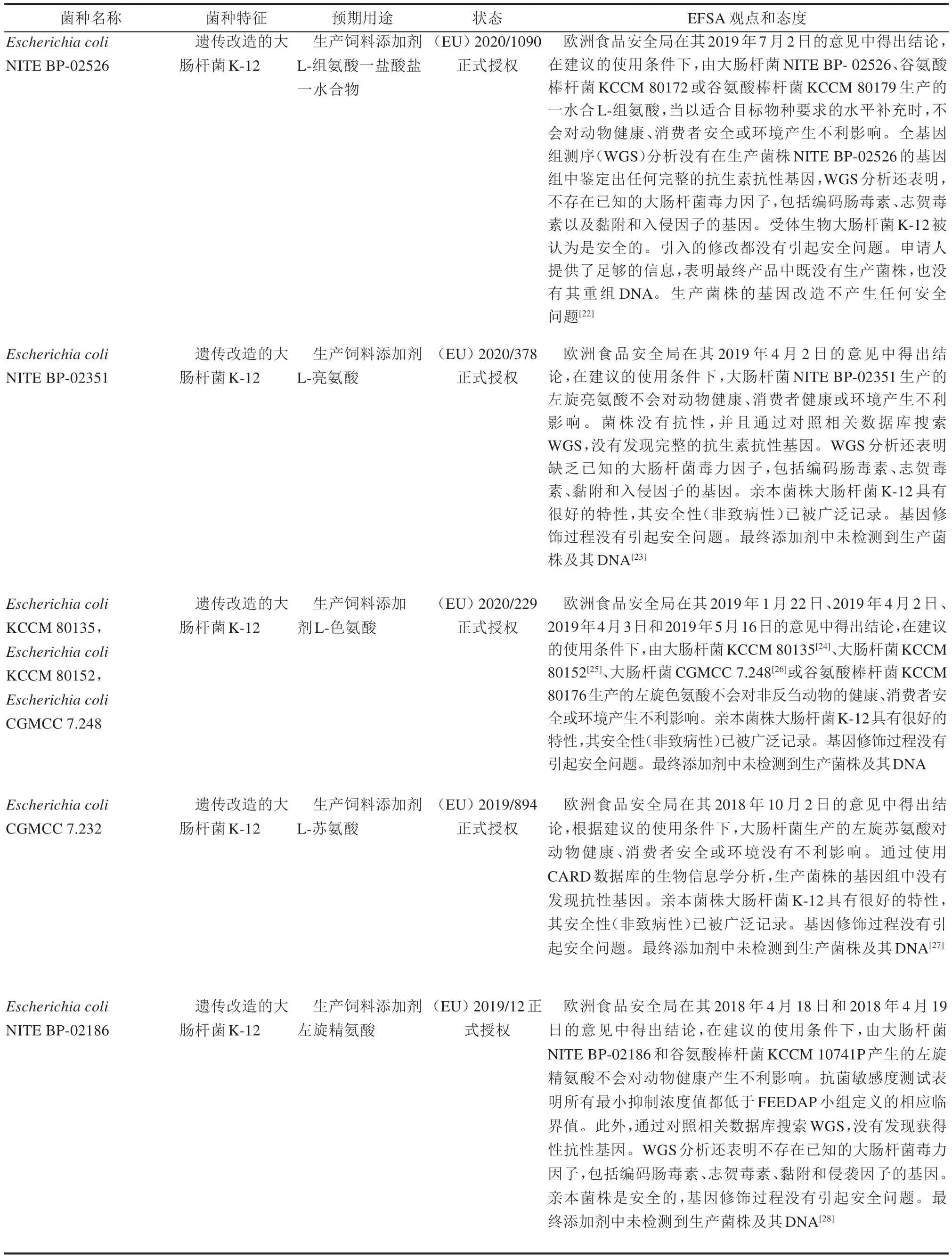
续表
6 结语与展望
合成生物学是发展最快、最有前途的新兴技术之一。合成生物学的迅速发展带来的各种转基因食品及产品日益影响着我们的社会生活,在带来巨大经济利益的同时,其中也潜藏着巨大的未知风险,需要法律加以规制。欧美是转基因技术发展最快也是技术水平最高的国家和地区,二者在转基因技术领域取得了辉煌的成就,然而因为二者的历史文化背景、技术水平和公众对转基因食品的认知程度不同,对转基因食品的监管也存在着截然不同的做法。其中以欧盟对转基因食品的监管最为严苛。鉴于欧盟市场的重要性,我们必须深入理解欧盟对于转基因食品的监管要求。广大科研工作者在设计遗传改造微生物时也必须要考虑相关监管法规的要求,从源头上减少欧盟准入成本、缩短周期。我国对转基因产品的界定仍然非常模糊,造成了“有技术、无市场”的尴尬局面,极大地限制了生物技术的应用与发展。我国现有的法规政策,难以适应转基因技术的发展,立法与制度建设严重滞后于科技发展。根据市场“法无禁止即可为”,同时,根据政府“法无授权不可为”,提出以下3 点建议。①合理借鉴其他国家的监管经验,参照欧美特别是欧洲的做法尽快推进相关法律法规出台,明确各类产品的申报、审批要求,统一市场准入标准,让相关产品的准入做到有法可依。②建议区分工业用遗传改造微生物和农业用遗传改造微生物,对于工业用遗传改造微生物的应用管理,可学习借鉴欧盟对遗传改造微生物制造食品、饲料的分类管理经验,对遗传改造微生物制造食品、饲料,按照终产品中是否检测出转基因组分等进行分类,对不同类别的产品采取不同的准入标准和措施;建议参照欧盟,建立我国的“QPS”清单,对于清单内的微生物制造产品简化评估内容和审批手续。③建议出台《中华人民共和国生物安全法》的配套准入注册指南,指导申请人完成准入注册。
猜你喜欢
区域治理(2022年40期)2022-11-27
学与玩(2022年10期)2022-11-23
今日农业(2022年3期)2022-06-05
理财·市场版(2020年7期)2020-08-06
动漫界·幼教365(小班)(2019年10期)2019-10-28
动漫界·幼教365(大班)(2019年10期)2019-10-28
动漫界·幼教365(中班)(2019年10期)2019-10-28
理财·市场版(2019年10期)2019-09-10
创新科技(2015年1期)2015-12-24
军事体育学报(2015年2期)2015-02-27
