
UPLC法同时测定阿托伐他汀钙中15个杂质含量
2021-03-16 04:56
食品与药品 2021年1期
(常州制药厂有限公司,江苏 常州 213000)
关健词:超高效液相色谱法;阿托伐他汀钙;有关物质
阿托伐他汀钙是HMG-CoA还原酶的选择性、竞争性抑制剂,能通过抑制肝脏内HMG-CoA还原酶和胆固醇的合成,降低血浆中胆固醇和脂蛋白水平;并通过增加肝细胞表面的低密度脂蛋白胆固醇(LDL-C)受体以增强LDL-C摄取和代谢。阿托伐他汀钙可降低纯合子型和杂合子型家族性高胆固醇血症、非家族性高胆固醇血症和混合型血脂异常患者的总胆固醇(TC),LDL-C和载脂蛋白B水平,为新一代他汀类降血脂药。
现有国内外产品质量标准、各国药典[1-3]及相关文献报道[4-12]中有关物质检测方法,存在分析时间过长、分离度较差等问题,均不能很好分离本文中涉及的15个杂质。经查阅文献,参考各国药典,采用灵敏度和分离度更高、分析速度更快的超高效液相色谱法(UPLC)测定阿托伐他汀钙中的杂质。本论文所建立的检测方法操作简便,结果准确,分离度与重复性好,分析效率高。
1 仪器与试药
1.1 仪器
岛津Nexera X2 LC-30AD超高压液相色谱仪,岛津Nexera X2 SPD-M30A光电二极管阵列检测器(日本岛津公司);Waters Empower 3软件(美国沃特世公司);XP6百万分之一电子天平,XP205DR十万分之一电子天平,FE20 pH计(瑞士梅特勒公司)。
1.2 试药
阿托伐他汀钙原料药(常州制药厂有限公司,批号:EAT190401,EAT190402,EAT190403,EAT190404,EAT191101,EAT191102,EAT191103,EAT191104);阿托伐他汀钙,杂质A、B、C、D对照品(USP);杂质M、O、P对照品(QCCUSA公司);杂质L、N对照品(Trc公司);杂质F、G、H、I、J、K(常州制药厂有限公司合成制备,经紫外、红外、质谱和核磁共振分析结构确证);乙腈,甲醇,N,N-二甲基甲酰胺(DMF),四氢呋喃(无稳定剂)为色谱纯;冰醋酸,醋酸铵为分析纯;试验用水为超纯水。
2 方法与结果
2.1 色谱条件
色谱柱:岛津Shim-pack Velox PFPP(2.1 mm×100 mm,1.8 μm);流动相A:3.9 g/L醋酸铵缓冲液(用冰醋酸调节pH至5.0):乙腈:甲醇:四氢呋喃(无稳定剂)=67:21:6:6,流动相B:3.9 g/L醋酸铵缓冲液(用冰醋酸调pH至5.0):乙腈:甲醇:四氢呋喃(无稳定剂)=27:61:6:6,按表1进行梯度洗脱,流速:0.43 ml/min;检测波长:244 nm;柱温:35 ℃;样品室温度:10 ℃;进样量:1.8 μl;稀释液:DMF。
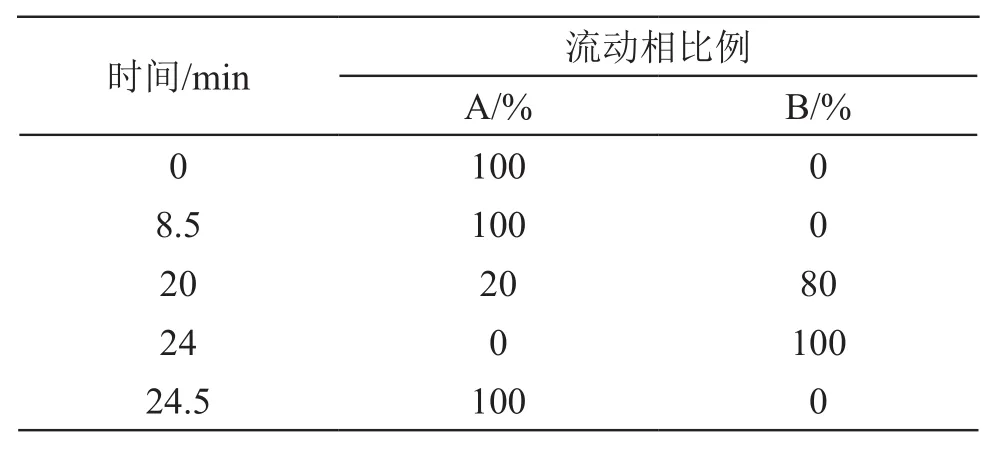
表1 流动相洗脱梯度
2.2 溶液的制备
杂质定位溶液:依次称取阿托伐他汀钙对照品、杂质A(阿托伐他汀脱氟物)、杂质B (阿托伐他汀非对映异构体)、杂质C (阿托伐他汀双氟物)、杂质D、杂质F(阿托伐他汀二聚物)、杂质G(阿托伐他汀甲酯)、杂质H(阿托伐他汀内酯)、 杂质I、杂质J(阿托伐他汀乙酯)、杂质K(阿托伐他汀叔丁酯)、杂质L、杂质M、杂质N、杂质O、杂质P对照品各5 mg,分别置入16个100 ml量瓶,加稀释液溶解稀释至刻度,摇匀。
系统适应性溶液:称取阿托伐他汀钙对照品和杂质A、B、C、D、F、G、H、I、J、K、L、M、N、O、P对照品各2.5 mg,置入同一50 ml量瓶,加稀释液溶解并稀释至刻度,摇匀。
标准曲线溶液:精密称取阿托伐他汀钙对照品和各杂质A、B、C、D、F、G、H、I、J、K、L、M、N、O、P对照品各5 mg,分别置入同一100 ml量瓶,加稀释液溶解稀释至刻度,摇匀,再精密移取5 ml至25 ml量瓶,加稀释液至刻度,摇匀作为线性贮备液,再分别移取0.3,0.5,1.0,1.5,2.0,2.5,3.0 ml线性贮备液,置入10 ml量瓶,加稀释液稀释至刻度,摇匀(相对于样品中杂质含量分别为0.03 %,0.05 %,0.10 %,0.15 %,0.20 %,0.25 %,0.30 %),作为线性溶液。
供试品溶液:称取阿托伐他汀钙样品约50 mg,置入50 ml量瓶,加稀释液溶解并稀释至刻度,摇匀。
对照溶液:量取1 ml供试品溶液,置入100 ml量瓶,加稀释液稀释至刻度,摇匀。再量取5 ml,置入50 ml量瓶,加稀释液稀释至刻度,摇匀。
降解实验溶液:称取阿托伐他汀钙样品7份,每份50 mg,分别置入50 ml量瓶,分别进行酸破坏(加0.5 mol/L盐酸溶液2 ml,放置24 h,加0.5 mol/L氢氧化钠溶液2 ml调pH至中性)、碱破坏(加0.5 mol/L氢氧化钠溶液4 ml,放置 24 h,加0.5 mol/L盐酸溶液4 ml调pH至中性)、氧化破坏(加30 %双氧水4 ml放置14 d)、光照破坏(置于4500±500 Lx下光照40 d)、高温破坏(置于105 ℃放置40 d)、高湿破坏(置相对湿度92.5 %放置40 d)和未进行破坏。取上述样品,分别加稀释液溶解稀释至刻度,摇匀作为供试品溶液;移取各破坏的供试品溶液1 ml,置入100 ml量瓶,加稀释液定容,摇匀;再移取5 ml,置入50 ml量瓶,加稀释液至刻度,摇匀,作为对照溶液;精密称取阿托伐他汀钙对照品25 mg,置入25 ml量瓶,加稀释液溶解,定容,摇匀,再精密吸取10 ml,置入25 ml量瓶,加稀释液溶解,定容,摇匀,作为含量对照品溶液(用于质量平衡);精密吸取各破坏的供试品溶液10 ml,置入25 ml量瓶,加稀释液稀释至刻度,作为含量供试品溶液。
2.3 结果
2.3.1 专属性试验 取系统适应性溶液和各杂质定位溶液,照2.1项下色谱条件进样分析,结果表明,本色谱条件下,阿托伐他汀钙主峰及各杂质峰间均能达到良好的分离,分离度均在1.5以上(见图1)。

AT:阿托伐他汀钙;IMP:杂质图1 系统适应性图谱注:杂质D和杂质M均出二个色谱峰
2.3.2 线性关系考察 取标准曲线溶液,照2.1项下色谱条件进样分析,由质量浓度(μg/ml)对峰面积做线性回归方程并计算其校正因子,具体数据见表2。结果表明,阿托伐他汀钙和各杂质均在0.3~3.0 μg/ml范围内线性良好。
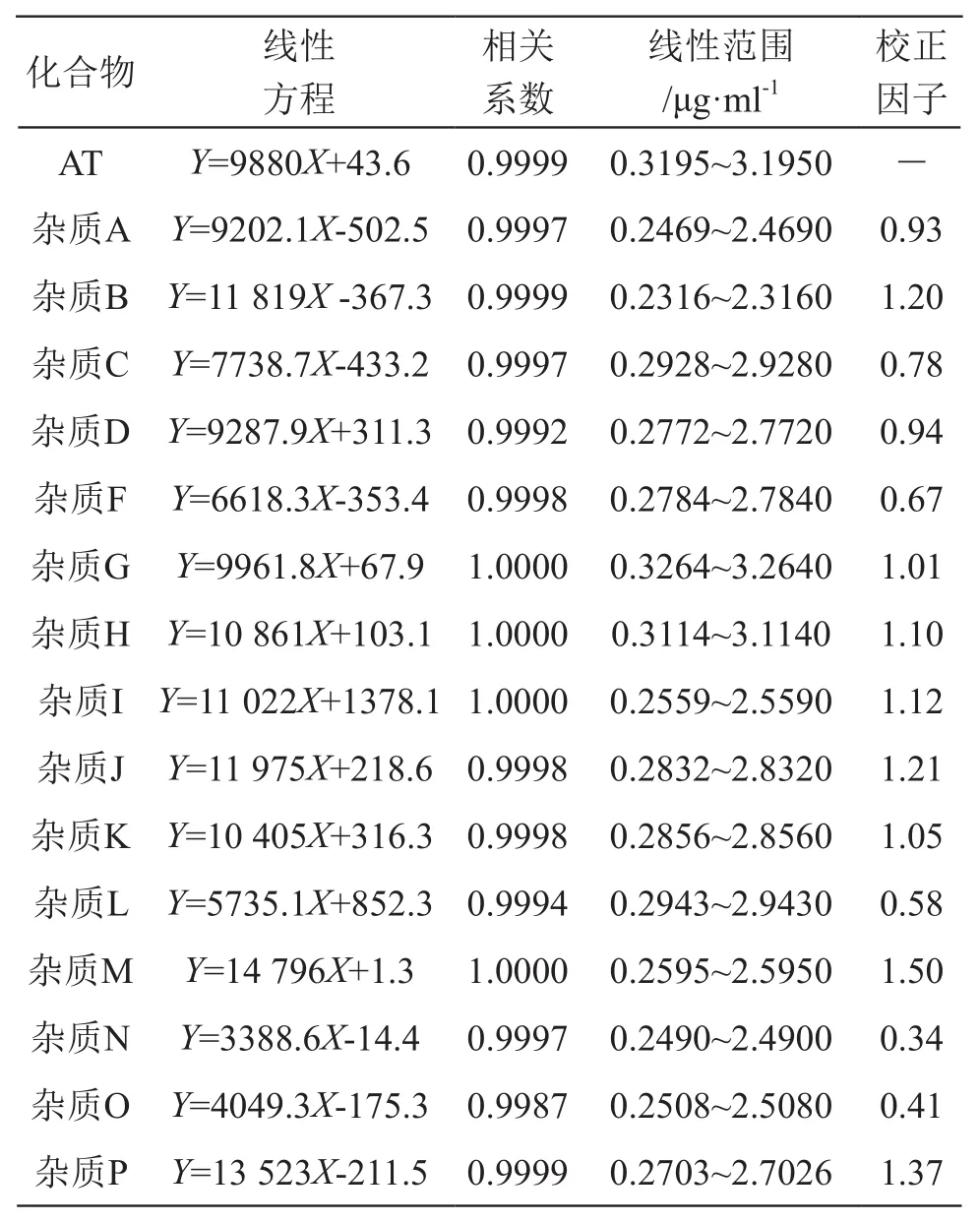
表2 线性数据
2.3.3 检测限和定量限 精密移取各杂质定位溶液适量,用稀释液逐级稀释,照2.1项下色谱条件进样分析,按信噪比S/N=10计算定量限,定量限溶液连续6针峰面积RSD均小于10 %,各杂质定量限浓度水平均小于0.03 %(相对于样品中杂质含量);按信噪比S/N=3计算检测限,各杂质检测限浓度水平均小于0.01 % (相对于样品中杂质含量),结果见表3。

表3 检测限和定量限
2.3.4 回收率试验 精密称取杂质A、B、C、D、F、G、H、I、J、K、L、M、N、O、P对照品各5 mg,分别置入同一100 ml量瓶,加稀释液溶解并稀释至刻度,摇匀,再精密移取10 ml,置入50 ml量瓶,加稀释液至刻度,摇匀,作为回收率贮备液,精密称取阿托伐他汀钙样品20 mg共12份,分别置入20 ml量瓶,再分别移取0.6,2.0,3.0,6.0 ml回收率贮备液置入20 ml量瓶,共3份,加稀释液稀释至刻度,摇匀(相对于样品中杂质含量分别为0.03 %,0.10 %,0.15 %,0.30 %),作为回收率供试品溶液。照2.1项下色谱条件测定上述12份溶液,计算每个杂质回收率和RSD,结果见表4,各杂质回收率均约100 %,且平行性较好。

表4 回收率试验结果(n=12)
2.3.5 重复性试验 称取阿托伐他汀钙样品20 mg,共6份,分别置入20 ml量瓶,再分别移取2.3.4项下回收率贮备液3.0 ml,置入20 ml量瓶,加稀释液溶解并稀释至刻度,摇匀(相对于样品中杂质含量为0.15 %)作为重复性试验供试品溶液,照2.1项下色谱条件,连续测定6份重复性试验供试品溶液,6份供试品溶液中各杂质含量RSD均小于5.0 %,结果表明,本方法本仪器重复性良好。
2.3.6 精密度试验
2.3.6.1 进样精密度试验 照2.1项下色谱条件,连续测定对照溶液6次,按峰面积计算RSD为0.1 %,结果表明,本方法本仪器进样精密度良好。
2.3.6.2 中间精密度试验 不同天,由另一名检验员,使用另一台UPLC仪器,按2.3.5项下方法配制6份中间精密度试验溶液,照2.1项下色谱条件,连续测定6份溶液,计算6个溶液中各杂质含量RSD均小于5.0 %。同时计算两名检验员间12份溶液各杂质含量RSD均小于5.0 %。结果表明,本方法中间精密度良好。
2.3.7 供试品溶液稳定性试验 称取阿托伐他汀钙样品约50 mg,置入50 ml量瓶,加稀释液溶解并稀释至刻度,摇匀作为供试品溶液,于仪器样品室中(10 ℃),分别于0,4,8,12,18,24,32,40,48 h,照2.1项下色谱条件进样分析,试验结果表明,供试品溶液48 h内较稳定(与初始值0 h比较,无新增杂质,无杂质消失,各杂质增减在0.05 %之内)。
2.3.8 耐用性考察 根据2.1项下色谱条件,采用不同品牌PFPP UPLC色谱柱、柱温(33,37 ℃)、流速(0.41,0.45 ml/min)、波长(242,246 nm)、流动相A中各组份比(66:22:6:6,68:20:6:6,67:21:5.8:6.2,67:21:6.2:5.8)和流动相B中各组份比(26:62:6:6,28:60:6:6,27:61:5.8:6.2,27:61:6.2:5.8),依次按2.1项下色谱条件进行系统适应性溶液试验。结果表明,不同品牌PFPP UPLC色谱柱保留时间差异较大,如不使用推荐色谱柱,可能造成杂质A、L、B及主峰分离不够;其他参数的微小变化下,各杂质峰间均能达到较好分离,基本不影响检测。上述结果表明除对色谱柱要求较高外,本方法耐用性较好。
2.3.9 降解试验结果 照2.1项下色谱条件依次进样降解试验溶液分析,结果见表5。在酸破坏、碱破坏、氧化破坏、高温破坏、光照破坏、高湿破坏等条件下,可检出除杂质A、杂质M、杂质G、杂质I除外的11个已知杂质,且阿托伐他汀钙主峰及检出各杂质峰的峰纯度均符合要求(峰纯度阈值大于峰纯度角值),且质量保持平衡。表明此法专属性好,能有效检测样品中的降解产物。
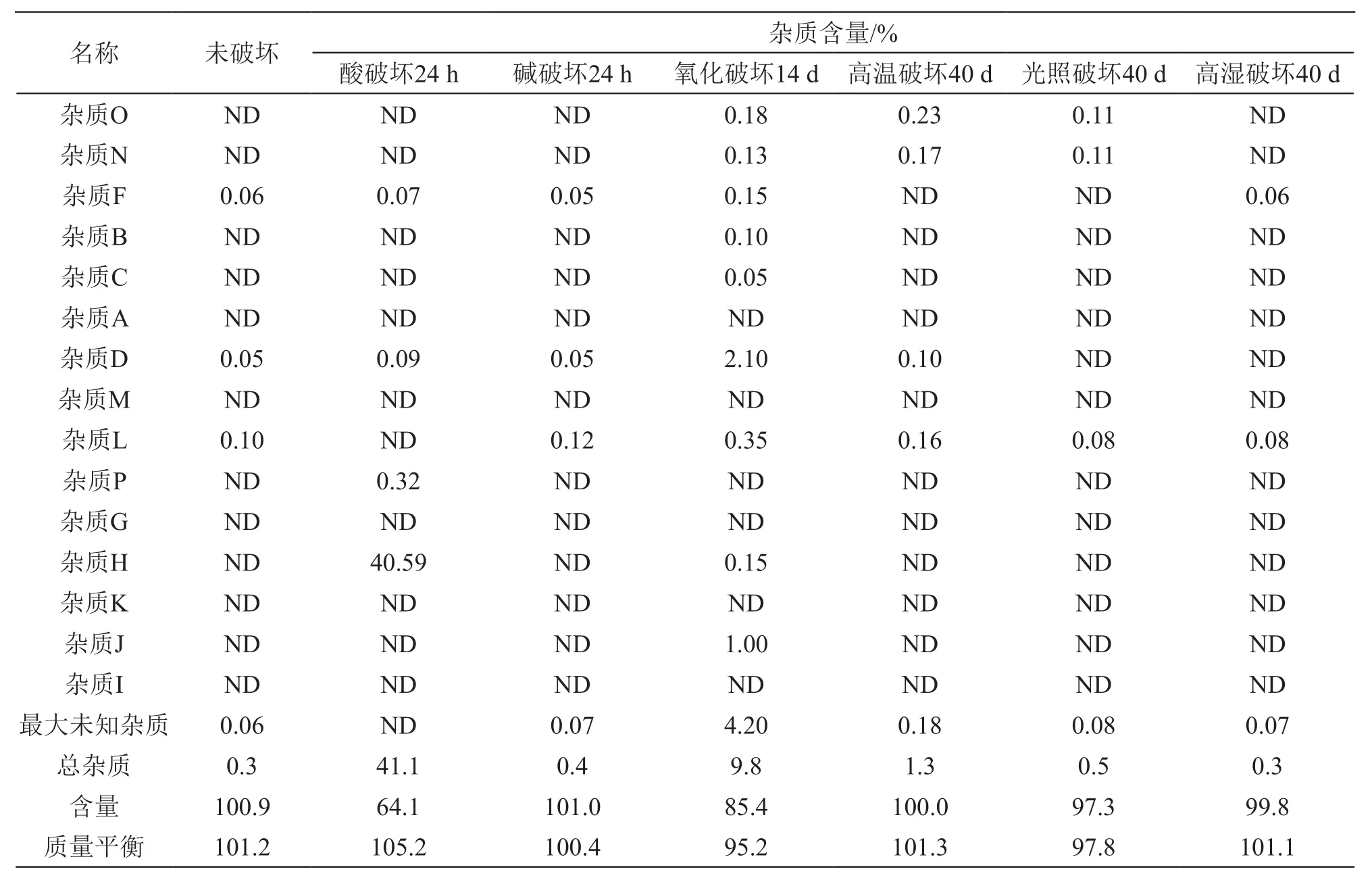
表5 降解试验数据
2.3.10 样品检测结果 对8个批次阿托伐他汀钙样品进行检测,结果见表6,第1~4批次样品均未检出文中研究的15个杂质,均检出未知杂质,杂质总量均在0.1 %左右,第5~8批次样品均检出杂质L,且第6~7批次样品均检出已知杂质F,但上述杂质含量均较低,单个杂质含量均低于0.1 %,杂质总量均低于0.15 %。

表6 8个批次样本检测数据
3 讨论
3.1 UPLC检测方法建立的必要性
检测上述15个已知杂质是各国药典以及相关企业内控质量标准的内在要求。在酸破坏、碱破坏、氧化破坏、高温破坏、光照破坏、高湿破坏等条件下,可检出除杂质A、杂质M、杂质G、杂质K、杂质I的10个已知杂质,上述破坏试验提示:在仓储、运输等过程中均有可能降解产生上述杂质,进而影响产品质量,因此建立简单、便捷、可定量的方法对控制产品质量有重要的实际价值。相较于传统HPLC方法,UPLC方法有分析时间更短、检测灵敏度更高,进样量更小等优势,更适应生产企业高效率检测等需要。
3.2 流动相选择
流动相选择参考了美国和欧洲药典收载流动相体系,并结合本实验室试验结果进行优化选择后最终确定。验证过程中发现,甲醇和四氢呋喃的用量对有些杂质之间分离影响较大,且四氢呋喃对基线梯度影响较大,因此需准确量取甲醇和四氢呋喃。四氢呋喃应不添加稳定剂,如含稳定剂会影响主峰前后的基线,影响杂质A,L,B检出能力及对照溶液主峰峰面积。同时要特别注意密封保存,防止水分及氧化物质进入使四氢呋喃变质,影响检验。
3.3 小结
本文以UPLC测定阿托伐他汀钙原料药中有关物质,结果表明,本法保留时间稳定,测定结果准确、精密度高、回收率符合检测要求。与常规液相色谱法比较,分析时间大幅缩短,分离效率高,重复性好,溶剂用量少,环保经济,可用于阿托伐他汀钙原料药的有关物质质量控制。
猜你喜欢
中国农业科技导报(2022年10期)2022-12-03
中国兽药杂志(2022年6期)2022-07-04
中国药学药品知识仓库(2022年13期)2022-07-03
医学食疗与健康(2022年2期)2022-04-23
中国药学药品知识仓库(2022年2期)2022-03-23
药品评价(2021年17期)2021-11-06
中国畜牧杂志(2021年3期)2021-03-15
黑龙江动物繁殖(2020年6期)2020-12-18
北方药学(2020年7期)2020-10-23
中国药业(2019年19期)2019-10-14
