
普瑞巴林的合成研究进展
2021-03-13 08:32:58朱元杰张兴光
合成化学 2021年2期
熊 非,陈 璐,朱元杰,马 超,张兴光
(上海理工大学 化学系,上海 200093)
普瑞巴林(Pregabalin,1,Chart 1),化学名为(S)-(+)-3-氨甲基-5-甲基己酸,是由美国辉瑞公司研发的一种新型γ-氨基丁酸(GABA)受体激动剂,并于2004年获得欧盟批准先后在英国和德国上市,商品名为Lyrica。它具有竞争性的与GABA受体结合产生抑制兴奋传导的作用,从而减少神经递质的释放,进而有效控制神经性疼痛,因此常用于治疗外周神经痛以及辅助性治疗局限性部分癫痫发作[1]。与临床上使用的加巴喷丁相比,普瑞巴林具有抗惊厥作用更强,不良反应更小,同时剂量低、服用次数少,且兼具抗焦虑作用,是加巴喷丁的升级换代产品,市场前景广阔。
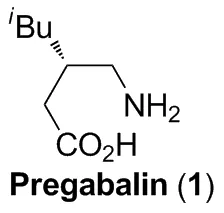
Chart 1
1的合成关键在于C-3位手性中心的构建,目前文献报道的合成策略主要分为外消旋体拆分法和不对称合成法。外消旋体拆分包括化学拆分、生物酶拆分和动力学拆分。化学拆分法是将合成的外消旋中间体与旋光性的酸或碱反应,再利用所形成非对映体盐之间的溶解性差异达到结晶拆分的目的。由于添加的旋光性拆分剂来源广泛,且可回收循环利用,具有潜在的工业应用价值。生物酶拆分法采用具有高度立体专一性的酶选择性对外消旋体中的一个对映体发生反应,从而使两个对映体分开,具有选择性好和拆分效率高等优势。动力学拆分法则是利用不足量的手性试剂与外消旋体作用,反应速度快的对映体优先完成反应,而剩下反应速度慢的对映体,从而达到拆分的目的。不对称合成是将潜手性结构单元转化为手性结构单元,从而直接产生不等量的立体异构产物,包括:1)手性底物控制的手性池合成法;2)手性辅基或手性试剂控制的手性辅助剂法;3)手性催化剂控制的不对称催化法。由于不对称合成法的合成效率较高,采用该合成策略对1的研究较为广泛。

Scheme 1
Scheme 2

Scheme 3
文献报道1的合成方法较多,本文根据不同的起始原料,及所采用的化学合成策略,综述和归纳了1及其重要中间体的合成路线。
1 中间体6的合成路线
以异戊醛(2)为起始原料的1的合成路线大多以3-异丁基戊二酸(5)或3-异丁基戊二酸酐(6)为关键中间体[2]。2与氰乙酸烷基酯在碱性条件下缩合制得3;3与丙二酸二乙酯进行共轭加成制得4;4再在酸性条件下水解脱羧得到二酸中间体5;或用氰乙酰胺与2进行先缩合再水解,两步反应生成5。然后使用脱水剂对5进行分子内脱水制得环状酸酐中间体6。
2 化学拆分合成策略
2.1 以3-异丁基戊二酸(5)为中间体的化学拆分合成路线
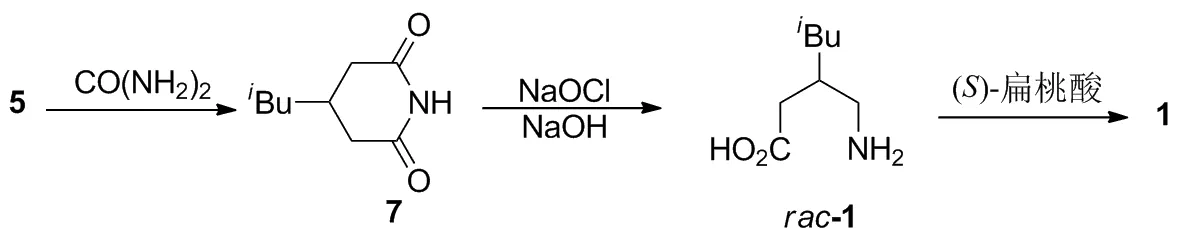
张贵森等[3]报道通过二酸5与尿素进行酰亚胺化反应,以90%的收率制得7;7在氢氧化钠作用下,先水解开环,再进行Hoffmann降解,以77%的收率制得rac-1;rac-1再用(S)-扁桃酸进行非对映体结晶拆分,以28%的分离收率得到(S)-1。该路线存在拆分率较低、拆分后剩余的(R)-1无法实现再利用等合成缺陷。
2.2 以3-异丁基戊二酸酐(6)为中间体的化学拆分合成路线
陈敖等[4]报道通过氨水对6进行胺解反应制得rac-8;然后对rac-8进行Hoffmann降解,以67%的收率得到rac-1;最后用(S)-扁桃酸进行非对映体结晶拆分,以35%的分离收率得到(S)-1。

Scheme 4
Scheme 5

Scheme 6

Scheme 7
为了解决拆分后剩余的(R)-1无法再利用的问题,Hoekstra等[5]采用相同的合成策略报道了一条先使用(R)-苯乙胺对rac-8进行结晶拆分得到光学活性的(S)-8,再对(S)-8进行Hoffmann降解制得(S)-1的合成路线。拆分后剩余的(R)-8可通过酰胺的水解重新形成酸酐6的合成前体二酸5,提高了该路线的原子经济性。
熊非等[6]报道了在三乙胺催化下对6进行醇解开环反应制得半酯rac-9,再用手性氯霉胺CHA对rac-9进行非对映体结晶拆分,以43%的分离收率得到光学活性的(S)-9,(S)-9先与氮化镁进行胺解反应,再经Hoffmann降解,以两步79%的收率制得1。拆分后剩余的(R)-9经过NaOH水解可重新形成酸酐6的合成前体二酸5,提高了原子经济性。另外,碱性拆分剂CHA作为工业生产氯霉素的副产物,价廉易得,让此路线具有潜在的工业化应用前景。
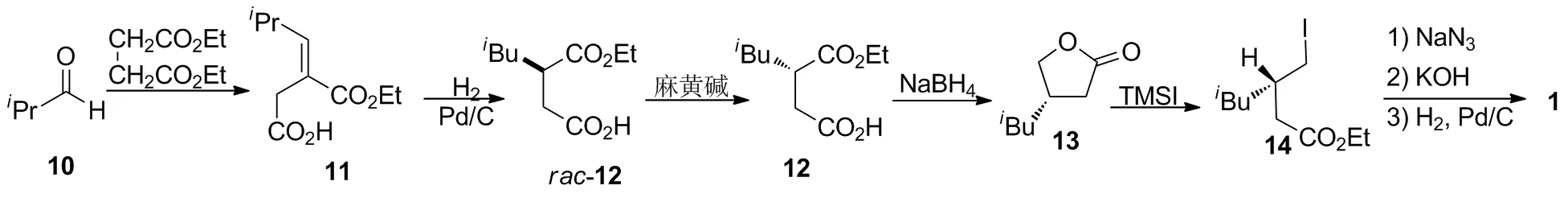
2.3 以异丁醛(10)为起始原料的化学拆分合成路线
Hoekstra等[5]报道使用10与丁二酸二乙酯进行Stobbe缩合,以90%的收率得到11;11在钯碳催化下进行氢化还原,以98%的收率得到rac-12;然后使用L-麻黄碱对rac-12进行非对映体结晶拆分,以46%的分离收率得到光学活性的(S)-12;12经过还原和内酯化一锅反应形成手性内酯13;13经过碘代开环得到14;再依次经叠氮化、水解和氢化还原,以3步61%的收率制得1。该路线中的关键步骤在于使用L-麻黄碱对rac-12进行非对映体结晶拆分,与(S)-扁桃酸和氯霉胺等拆分剂相比,麻黄碱作为易制毒化学试剂不易获取。此外,该路线还存在合成步骤冗长,总收率不高等缺陷。
2.4 以异戊醛(2)为起始原料的化学拆分合成路线
Hoekstra等[5]在二正丙胺存在的条件下,将2与丙二酸二乙酯进行缩合,以89%的收率得到α,β-不饱和二酯15;15与氰化钾进行共轭加成,以94%的收率形成rac-16;rac-16再依次经水解脱羧和加氢还原,以73%的收率(以rac-16计)转化为rac-1;最后采用(S)-扁桃酸进行化学拆分,以29%的收率得到1。
Ishitani等[7]采用非均相催化的管式连续流反应装置将2和丙二酸二甲酯通过Knoevenagel缩合反应转化为17,然后与硝基甲烷进行共轭加成,再经过氢化还原和分子内的胺解,以几乎定量的收率制得rac-18;rac-18依次在NaOH和HCl溶液中进行水解、脱羧和中和,以两步67%的收率制得rac-1。与传统均相反应相比,该路线在制备中间体rac-18的过程中采用非均相催化管式连续流反应,更环保、安全,但该路线仍存在拆分rac-1后剩余的(R)-1无法实现再利用的问题。
Scheme 8

Scheme 9
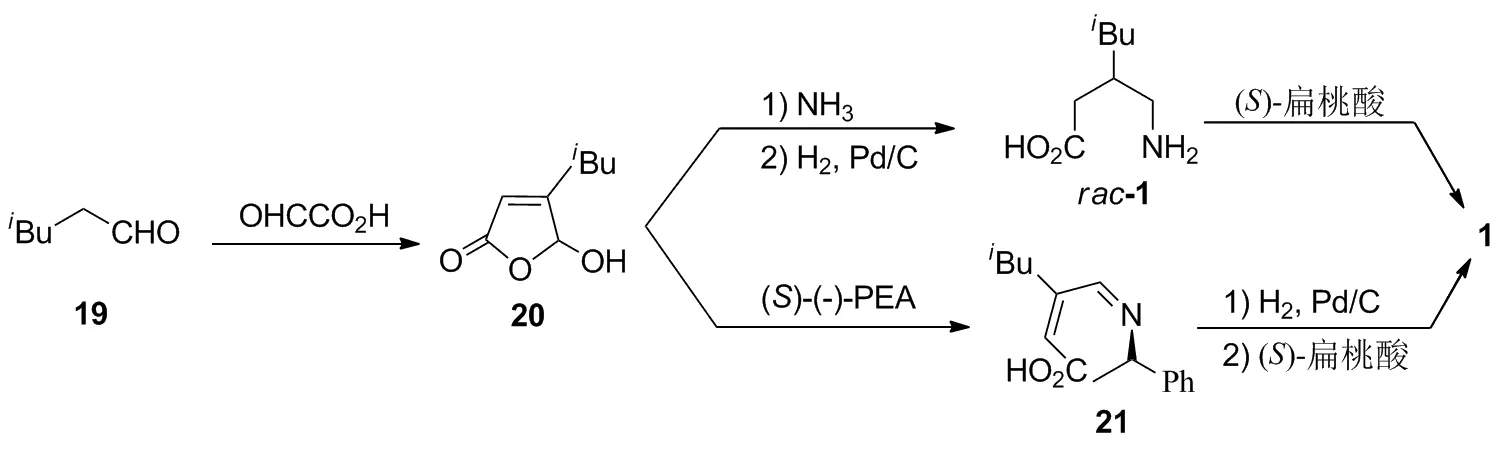
2.5 以4-甲基戊醛(19)为起始原料的合成路线
Roy等[8]报道将19与乙醛酸反应,以60%的收率得到20;20再依次与氨气进行缩合、钯碳催化氢化后得到rac-1,最后用(S)-扁桃酸进行结晶拆分,以61%的收率(以20计)制得(S)-1。以20为共同合成中间体,他们还报道了另外一条20与(S)-苯乙胺反应,以93%的收率转化为亚胺21;21再依次经催化氢化和使用(S)-扁桃酸进行结晶拆分[9]制得1的路线。上述合成路线虽然步骤短,但依然存在(S)-扁桃酸拆分rac-1后剩余的(R)-1无法实现再利用,原子经济性较低等缺陷。
2.6 以4-甲基戊腈(22)为起始原料的化学拆分合成路线
Shelke等[10]将22与氯苄在LDA作用下发生亲核取代,以80%的收率形成23;再采用“氧化-还原”合成策略,即对23先用高碘酸钠选择性氧化苄基,以70%的收率得到rac-24,然后再用硼氢化钠还原氰基,以88%的收率制得rac-1。同时他们还报道了另外一条以23为关键中间体的合成路线,并采用了“还原-氧化”合成策略:即先用硼氢化钠和BOC酸酐与23进行一锅法反应,以85%的收率转化为25,再用高碘酸钠选择性氧化,以76%的收率得到26,然后在盐酸作用下水解脱除胺基保护基,以88%的收率制得rac-1。两条合成路线依然存在拆分rac-1后剩余的(R)-1无法实现再利用,原子经济性较低的问题。另外,在第二条合成路线中为了避免还原产物中胺基被氧化,需要增加胺基的保护和脱保护的合成步骤,降低了总收率。
2.7 以丙二酸二乙酯衍生物为起始原料的化学拆分合成路线
Bobade等[11]将二酯27在碱性条件下水解转化为二酸28;28再与二乙胺、甲醛发生Mannich反应得到29;29依次经乙酯化、氢化铝锂还原、磺酯化、叠氮化等4步反应,以46%的收率(以29计)得到31;31再依次经还原、酰胺化和BOC保护后,以47%的收率制得32;32再在Grubbs催化剂的作用下,发生烯烃复分解反应,以50%的收率生成33,然后经硼氢化钠还原、三氟乙酸脱保护和盐酸作用下的水解开环反应,以53%的收率得到rac-1。然而上述合成路线依然存在拆分rac-1后剩余的(R)-1无法实现再利用,原子经济性较低的问题。同时该路线合成步骤冗长,缺乏工业化竞争力。

Scheme 10

Scheme 11
Lima等[12]使用Boc酸酐保护的氨基硼酸酯35作为α-氨基自由基源,在Lewis碱DMAP和光催化剂Mes-Acr-4存在的反应条件下,对二酯34进行共轭加成,以86%的收率得到36;再在酸性条件下对36进行一锅法水解、脱保护、脱羧,以94%的收率得到rac-1。该路线采用光催化合成策略,只需3步反应即可制备rac-1,然而依然存在拆分rac-1后剩余的(R)-1无法实现再利用的问题。
Yamada等[13]以34与N-碘甲基邻苯二甲酰亚胺(37)为原料,在叔丁基过氧化氢(TBHP)和二乙基锌的作用下发生自由基型共轭加成,以84%的收率转化为38;然后对38进行水解和酸性条件下回流脱羧,以两步69%的收率制得rac-1。该路线中过氧化物TBHP对光敏感,稳定性差,不利于生产操作。Chu等[14]在可见光作用下,使用铱催化剂Ir[dF(CF3)ppy]2(dtbbpy)+对34与2-叔丁氧羰基氨基乙酸发生自由基型共轭加成反应,以96%的收率得到39;39再依次发生脱除Boc基团保护和水解脱羧反应,以两步59%的收率制得rac-1。该路线采用可见光催化的光致氧化还原为关键反应,避免了使用对光和热敏感的过氧化物作为自由基引发剂,具有简单高效、绿色环保等特点,然而贵金属铱催化剂的来源和制备、拆分rac-1后剩余的(R)-1无法实现再利用等问题限制了其工业应用。
2.8 以重氮化合物40为起始原料的化学拆分合成路线
Chen等[15]在Rh2(OAc)4催化作用下,对40进行分子内的碳氢键插入反应,以91%的收率转化为内酰胺41;然后在三氟乙酸作用下脱除异丙苯保护基,以87%的收率得到rac-42;再在盐酸作用下水解开环,以90%的收率制得rac-1。该路线虽然总收率较高,但需要使用价昂的贵金属铑催化剂和易爆炸的重氮化合物。此外,还存在拆分rac-1后剩余的(R)-1无法实现再利用等缺陷。
2.9 以2,4-二甲基-1-戊胺(43)为起始原料的化学拆分合成路线
Wang等[16]将烷基胺43与2-氯甲酰基吡啶进行酰胺化反应,以84%的收率得到44;然后使用四甲基哌啶氮氧化物TEMPO作为氧化剂选择性对44中sp3杂化的碳氢键进行钯催化的羰基化反应,以80%的收率得到内酰胺45;再在盐酸溶液中对45进行水解开环,以86%的收率制得rac-1。该路线以碳氢键活化官能团化反应为核心,合成步骤较短,但需要使用价昂的贵金属钯催化剂,同时还存在拆分rac-1后剩余的(R)-1无法实现再利用的问题,不具有工业化应用的优势。
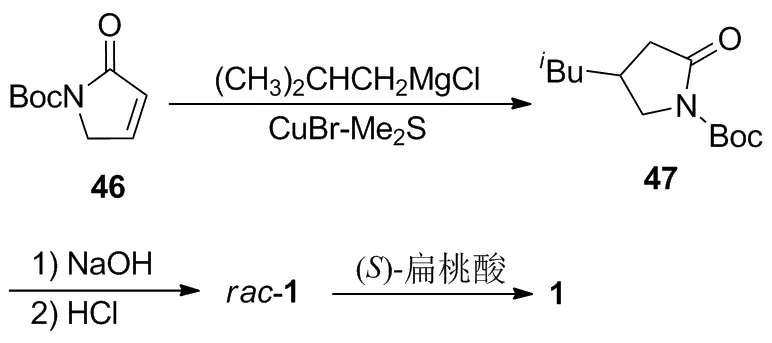
2.10 以α,β-不饱和内酰胺46为起始原料的化学拆分合成路线
郝二军等[17]在CuBr-Me2S催化作用下,将α,β-不饱和内酰胺46与异丁基格氏试剂进行共轭加成,以80%的收率得到47;47无需纯化,再依次在NaOH和HCl溶液中水解、脱保护,以95%的收率得到rac-1;最后用(S)-扁桃酸对rac-1进行非对映体结晶拆分,以55%的收率得到(S)-1。该路线较为简捷,遗憾的是需要使用稳定性较差的格氏试剂作为反应试剂。

Scheme 12

Scheme 13

Scheme 14
Scheme 15
2.11 以乙烯基乙酰胺衍生物48为起始原料的化学拆分合成路线
Liu等[18]使用醋酸钯催化48与邻苯二甲酰亚胺和2-甲基-1-碘丙烯的三组分碳铵化反应,以65%的收率得到49;然后对49进行BOC酸酐活化、酰胺水解一锅法脱除8-氨基喹啉导向基,以97%的收率得到50;再对50进行氢化还原、盐酸水解脱保护,以75%的收率制得rac-1。该路线需要使用贵金属钯催化剂,同时还存在拆分rac-1后剩余的(R)-1无法实现再利用的问题。
3 酶拆分合成策略
3.1 以异丁醛(10)为起始原料的酶拆分合成路线
Roy等[8]将10与丁二酸二苄酯进行缩合,以66%的收率得到51;对51进行酯化和氢化还原后,以两步55%的收率得到52;52依次与二氯亚砜、氨气进行酰胺化,再在二氯亚砜中脱水,以3步69%的收率得到rac-53;然后用南极假丝酵母脂肪酶Novozym 435对rac-53进行酶拆分,以50%的收率和99%ee值得到光学活性的(S)-53;在LiOH作用下对(S)-53进行水解,以71%的收率得到54;54经还原和碱化,以60%的收率得到1。酶拆分的立体选择性虽好,但拆分剂易失活且稳定性差。
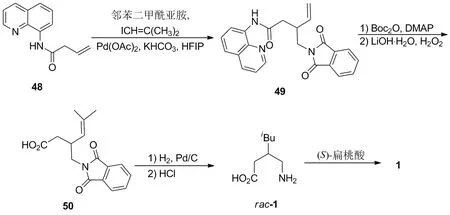
3.2 以异戊醛(2)为起始原料的酶拆分合成路线
在二正丙胺[19]或哌啶[20]存在的条件下,2与丙二酸二乙酯进行缩合得到α,β-不饱和二酯15,15再与氰化钾进行共轭加成形成β-氰基酯rac-16。Martinez[19]和Mukherjee[20]等分别使用Lipolase和Lipase酶对rac-16进行酶拆分得到光学活性的(S)-16,再经过水解脱羧、氢化还原制得光学活性的(S)-1。作者还报道了可以将拆分后剩余的(R)-16进行消旋化重新形成rac-16的方法,提高了路线的原子经济性,具有潜在的工业化应用价值。
Scheme 16

Scheme 17
Scheme 18

Scheme 19
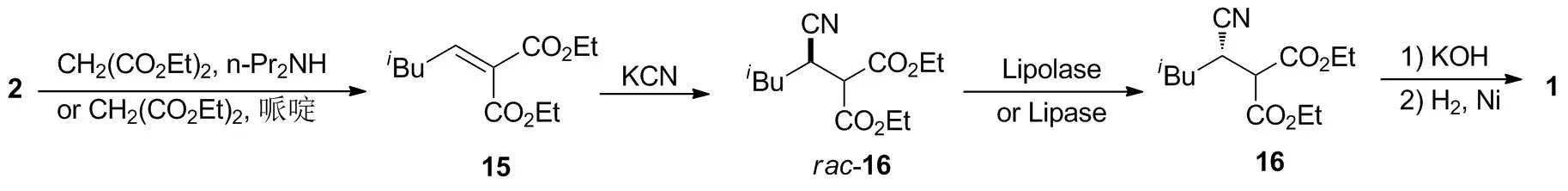
Felluga等[21]将2与二乙氧基膦酰基乙酸乙酯缩合,以80%的收率得到α,β-不饱和酯55;然后在DBU作用下与硝基甲烷共轭加成,以80%的收率得到rac-56;rac-56在Novozym 435酶作用下选择性酶解拆分,以21%的拆分率和92%的ee值得到(S)-56;(S)-56经镍催化氢化制得1。该路线尽管拆分产物ee值较高,但拆分率偏低。
4 动力学拆分合成策略
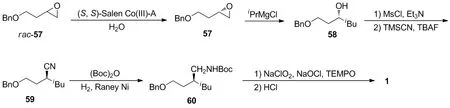
4.1 以环氧化合物57为起始原料的化学动力学拆分合成路线
Mujahid等[22]在手性Jacobsen催化剂[(S,S)-Salen Co(III)-A]的作用下,对rac-57进行水解动力学拆分,以40%的柱层析分离收率得到光学活性的(S)-57;然后在异丙基格氏试剂的作用下对57进行亲核开环,以90%的收率得到58;58先在甲磺酰氯作用下进行磺酯化,再进行氰基化取代,以两步71%的收率得到59;59再依次进行镍催化的氢化还原、BOC酸酐保护、亚氯酸钠氧化和盐酸作用下的水解脱保护,以69%的收率(以59计)制得1。该路线的合成缺陷是需要使用稳定性较差的格氏试剂作为反应试剂。
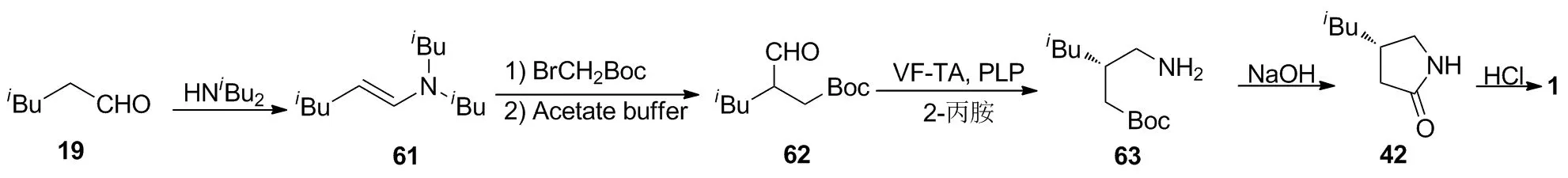
4.2 以4-甲基戊醛(19)为起始原料的生物动力学拆分合成路线
Fuchs等[23]将19与二异丁胺进行缩合得到烯胺61;61再与溴乙酸叔丁酯进行烷基化反应,以20%的收率(以19计)转化为62;然后在生物转氨酶VF-TA作用下与2-丙胺发生不对称胺化,以55%收率和60%ee得到光学活性的(S)-63;63先在碱性条件下发生酯基的分子内胺解,以70%的收率和90%ee得到內酰胺(S)-42;(S)-42再在盐酸中水解开环,以63%的收率制得1。生物酶具有无毒、污染小,催化副反应少、收率高等特性,是理想的动力学拆分试剂,但该路线拆分所得手性中间体的光学纯度还有待进一步提高。
Scheme 20
Scheme 21

Scheme 22
Scheme 23
5 手性池合成策略
5.1 以甘露醇缩二丙酮(64)为起始原料的手性源合成路线
Izquierdo等[24]使用高碘酸钠对手性起始原料64进行选择性氧化,以93%的收率制得65;65与二乙氧基膦酰基乙酸乙酯进行缩合,以80%的收率得到66;66在四丁基氟化铵(TBAF)作用下与硝基甲烷进行共轭加成,以75%的收率得到67;然后67在氢氧化钯催化的甲酸铵转移氢化反应中,以85%的收率转化成68;68再依次经BOC酸酐保护胺基、缩酮水解反应,以定量的收率形成二醇中间体69;69经高碘酸钠选择性氧化,以定量的收率得到70;70先与wittig试剂进行缩合,再在氢氧化锂溶液中水解开环,以60%的收率生成71;最后71在钯催化下脱除BOC保护,以定量的收率制得1。该路线合成步骤过于冗繁,无工业化应用价值。
Scheme 24

Scheme 25
Scheme 26
Scheme 27
5.2 以γ-丁内酯72为起始原料的手性源合成路线
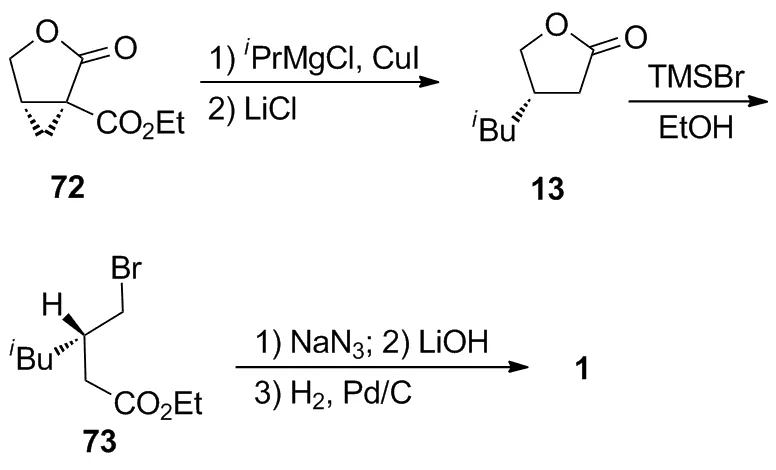
Ok等[25]使用异丙基格氏试剂对72进行亲核开环反应和氯化锂作用下的去乙酯化反应,以两步75%的收率生成13;13再依次进行溴代、叠氮化、酯基水解和叠氮基还原,以4步76%的收率制得1。该路线虽然合成步骤较短,但由于手性原料72来源不广泛和需要使用危险性试剂叠氮化钠,在工业化应用中无太大竞争力。
5.3 以L-亮氨酸(74)为起始原料的手性源合成路线
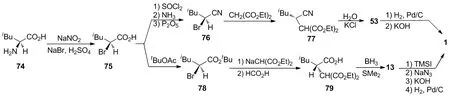
Roy等[8]报道了将74与亚硝酸钠、溴化钠和硫酸反应,以85%的收率得到75;75再依次经酰氯化、酰胺化和脱水反应,以3步72%的收率转化为76;然后76与丙二酸二乙酯进行亲核取代得到77;77再水解脱羧,以两步79%的收率和60%的ee值得到53;53再依次经氰基的氢化还原、酯基的碱性水解得到1。该路线中手性中间体的光学纯度较差,且大多为油状物,不利于产物的分离。Hoekstra等[5]报道了另外一条以75为关键手性中间体的立体专一性全合成路线:将75与乙酸叔丁基酯进行酯交换,以83%的收率得到78;78与丙二酸二乙酯的钠盐进行亲核取代,再用甲酸对叔丁酯进行选择性水解,以两步93%的收率得到79;然后用硼烷和甲硫醚进行脱羧还原和内酯化一锅反应,将79转化为13;13再与三甲基碘硅烷进行开环反应、叠氮化、水解和氢化还原,以57%的收率(以79计)制得1。该路线合成步骤冗长,并且还需要使用危险性试剂叠氮化钠。

Scheme 28

Scheme 29
Scheme 30

Scheme 31
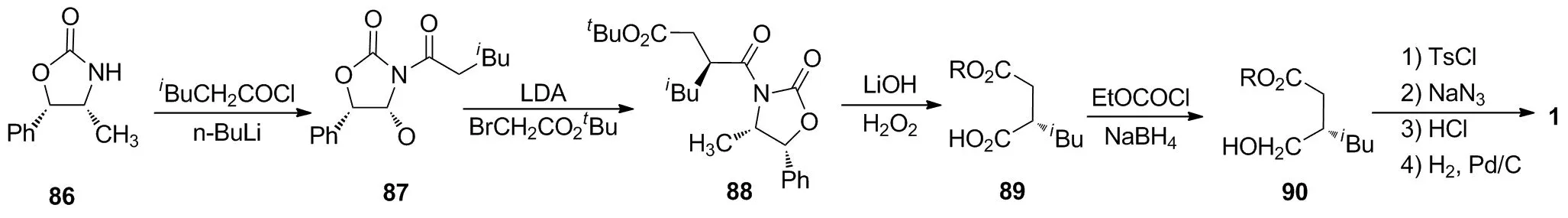
5.4 以(S)-3-氨甲酰甲基-5-甲基己酸(8)为起始原料的手性源合成路线
姚成志等[26]将8在氢氧化钠的水溶液中与NaClO先进行氧化反应得到含有80的反应液,然后在反应液中加入还原剂Na2SO2得到含有81的反应液,最后依次进行催化氢化还原反应液中形成的副产物、酸化和重结晶,以85%的收率制得1。该路线在氧化步骤后加入还原剂破坏残留的氧化剂,有效的降低了形成含有不饱和烯烃官能团的副产物产生,同时也巧妙利用了催化氢化将形成的微量含不饱和官能团的副产物还原转化为1,提高了原子经济性。此外,该路线还具有反应条件温和、中间体无需分离、适合一锅法进行等特点,适合工业化应用。
6 手性辅助剂合成策略
6.1 以(R)-4-苯基-2-恶唑烷酮(83)为手性辅助剂的合成路线
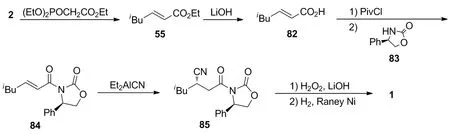
Tovar等[27]以异戊醛(2)为起始原料与二乙氧基膦酰基乙酸乙酯进行缩合,以86%的收率形成55;55在氢氧化锂作用下水解,以89%的收率得到82;用三甲基乙酰氯对82进行酰化后,再与手性辅助剂83进行酰胺化反应,以两步62%的收率制得84;84与二烷基氰化铝进行非对映选择性共轭加成,以57%的收率和74%的de值得到85;最后依次进行水解和氢化还原,以两步95%的收率制得1。该路线通过在反应底物中引入手性辅基控制共轭加成产物的非对映选择性,但存在不对称合成的产物de值不高,且需要通过柱层析分离等缺陷。
6.2 以(4R,5S)-4-甲基-5-苯基-2-恶唑烷酮(86)为手性辅助剂的合成路线
Hoekstra等[5]将86在正丁基锂作用下与4-甲基戊酰氯进行酰胺化,以94%的收率形成87;87在LDA作用下与溴乙酸叔丁酯进行立体选择性烷基化偶联,以74%的收率得到88;88在氢氧化锂和双氧水的作用下水解脱除手性辅基,以94%的收率得到89,同时以94%的回收率回收了手性配体86;89经硼氢化钠和氯甲酸乙酯还原,以几乎定量的收率得到90;90再经磺酯化、叠氮化、酯基水解和氢化还原后,以4步48%的收率和99%ee制得1。与采用手性配体83的合成路线相比,该路线大幅提高了手性中间体和产物的光学纯度,同时也对手性配体的回收再利用进行了研究,但合成步骤相对较长、需要使用危险性试剂叠氮化钠等因素限制了其工业化应用。
6.3 以手性α-苯乙胺91为手性辅助剂的合成路线
Rodríguez等[28]将(R)-91与甲酰化膦盐进行亲核取代反应制得92;92再与异丁醛进行Wittig反应,以72%的收率(以91计)生成93;93与溴代乙酰溴进行酰胺化反应,以92%的收率得到94;然后在三正丁基氢化锡的催化作用下,对94进行自由基环加成,以76%的收率得到95;95再经过还原脱除手性辅基和水解开环,以68%的收率制得1。
竺伟等[29]以3-异丁基戊二酸(5)为起始原料,在乙酸酐中发生分子内脱水,以定量的收率形成环状酸酐6;然后用(R)-91对6进行不对称胺解,以81%的收率和99%的de值制得关键手性中间体96;最后先对96进行催化氢化脱除手性辅基,再通过Hoffmann降解和pH调节,以3步74%的收率制得1。他们[30]同时还报道了将5与尿素反应,以92%的收率得到酰亚胺7;然后用(S)-91对7进行不对称胺解,以90%的收率和99%的de值制得关键手性中间体97;最后先对97进行Hoffmann降解,再对97进行酰胺的水解脱除手性辅基和pH调节,以3步87%的收率制得1。该路线反应步骤较少,各步收率和关键反应的非对映选择性高,具有良好的工业化应用前景。
7 不对称催化合成策略
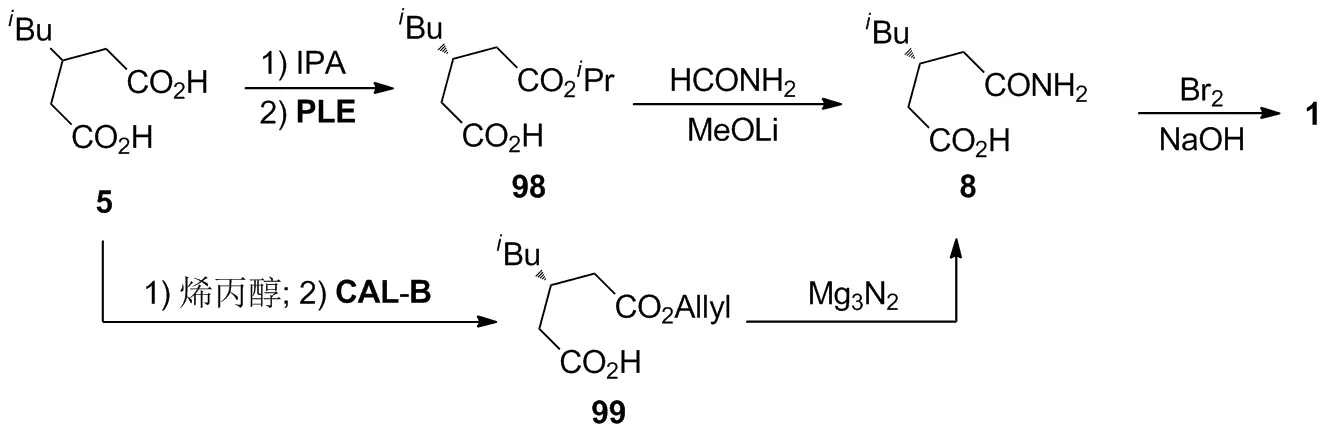
7.1 以3-异丁基戊二酸(5)为起始原料的酶催化合成路线
Hoekstra等[5]将5与异丙醇进行酯化,再通过PLE酶进行酯基的选择性催化水解,以两步82%的收率和85%的ee值制得光学活性的98;98与甲酰胺和甲醇锂反应,以66%的收率制得8;8再经Hoffmann降解制得1。Jung等[31]报道5与烯丙醇先进行酯化,再采用CAL-B酶进行酯基的选择性催化水解,以99%的收率和93%的ee值制得99;99与氮化镁进行胺解,以93%的收率得到8;8经Hoffmann降解,以75%的收率制得1。采用酶催化的不对称合成策略可以实现1的高对映选择性合成,但酶催化剂价昂,并且对温度和pH敏感,限制了其进一步的工业化应用。
7.2 以3-异丙基戊二腈(100)为起始原料的酶催化合成路线
Duan等[32]使用氰基水解酶HsNIT对100进行对映选择性催化水解,以93%的收率和94%的ee值得到101;101在DPPA和甲醇作用下,经Curtius重排反应,以48%的收率得到102;最后在盐酸溶液中对102进行回流水解,以73%的收率和89%的ee值制得1。该路线合成步骤较短,但中间体及产物的光学纯度较差,以及氰基水解酶的来源和价格等因素限制了其进一步的工业化应用。
7.3 以3-异丁基戊二酸酐(6)为起始原料的不对称催化合成路线
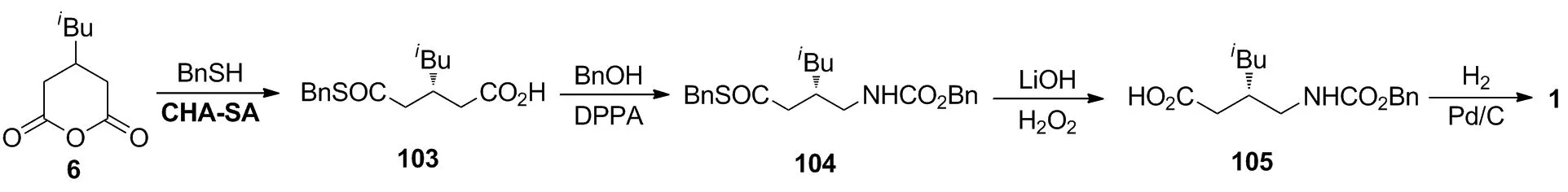
杨鸿均等[33]报道将苄硫醇与6在氯霉胺衍生的手性磺酰胺催化剂CHA-SA催化作用下进行不对称醇解,以89%的收率和94%的ee值制得103;103先与DPPA和苄醇进行curtius重排,再与氢氧化锂作用,以两步71%的收率转化为105;105经催化氢化脱保护,以60%的收率制得终产物1。
Scheme 32

Scheme 33

Scheme 34

Scheme 35
Scheme 36
Park等[34]将苄醇与6在奎宁衍生的手性磺酰胺催化剂QN-SA催化作用下进行不对称催化醇解,以 90%的ee值得到106;106经Curtius重排后,再进行催化加氢脱除保护基,以两步87%的收率制得1。上述两条路线均采用对6的不对称醇解开环反应为关键步骤,区别在于所采用的磺酰胺类催化剂的手性母体结构不同,与奎宁相比,氯霉胺作为工业生产氯霉素的废弃副产物,更廉价易得。但它们均需进行催化剂母体结构的改造,且制备过程步骤繁琐,部分中间体还需要经过柱层析进行纯化处理,不利于工业化应用。
Hamersak等[35]用反式肉桂醇与6在奎宁催化作用下进行不对称醇解,以72%的收率和97%的ee值制得(R)-9;(R)-9再依次经Curtius重排、醋酸钯和三苯基膦作用下的酯基水解、钯催化下脱除氨基保护基,以3步52.8%的收率制得1。奎宁催化剂作为天然金鸡纳生物碱,具有来源广泛、无需进行结构修饰等优势,但中间体需要经过柱层析进行纯化处理。
7.4 以异丁醛(10)为起始原料的不对称催化合成路线
Burk等[36]将10与氰乙烯在DABCO和BHT的作用下发生亲核加成,以97%的收率形成108;然后在吡啶作用下,与氯甲酸乙酯进行酯化,以95%的收率得到109;109经醋酸钯催化的羰基化,以83%的收率制得110;再用叔丁醇胺对110进行酯基的胺解,以89%的收率得到111;在贵金属铑的络合物催化作用下对111进行氢化还原,以100%的转化率和97.7%ee值制得112;最后依次进行氰基的氢化还原、酸性水解反应,以两步61%的收率制得1。然而该路线需要使用价昂的贵金属铑催化剂。

Scheme 37

Scheme 38
Scheme 39

Scheme 40
7.5 以异戊醛(2)为起始原料的不对称催化合成路线
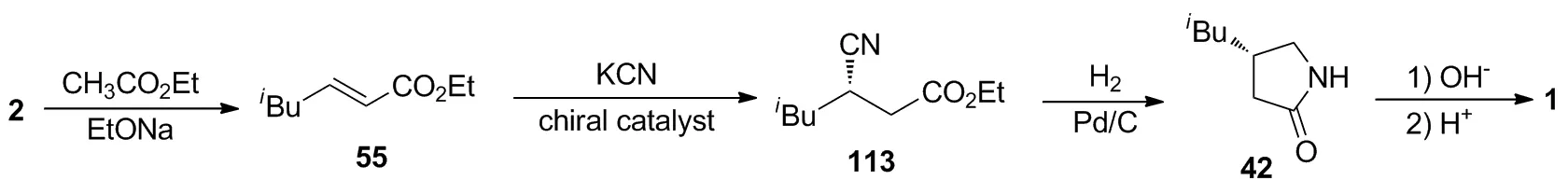
方向等[37]将2在碱性条件下与乙酸乙酯缩合,以90%的收率制得55;55与氰化钾在手性相转移催化剂1-(9-蒽甲基)-((R)-(6-羟基-4-喹啉基)-苯氧甲基-2-基)-5-乙基-1-氮杂双环[2.2.2]辛烷的催化作用下进行不对称迈克尔加成,以93%的收率和99.7%ee值得到(S)-113;对113进行氢化还原和环化一锅法制得42,再在碱性条件下对42进行水解开环,最后通过溶液pH的调节,以92%的收率(以113计)得到产物1。该路线采用不对称共轭加成为关键反应实现了高对映选择性和高收率的制备1,具有原料价廉易得、反应步骤少、收率高等优势,适合工业化生产。
Leyva-Perez等[38]将2与硝基甲烷和丙二酸二乙酯在固载于介孔硅酸材料的手性脲催化作用下进行三组份串联反应,以31%的收率和81%的ee值制得114;再对114进行氢化还原和内酰胺化,一锅法得到42,最后42在盐酸中水解开环制得1。该路线中尽管手性中间体114的ee值较高,但该步反应的收率偏低。Liu等[39]使用环己二胺衍生的手性硫脲催化α,β-不饱和硝化物115与丙二酸二乙酯的不对称共轭加成,以73%的收率和88%的ee值制得114;对114进行氢化还原,以72%的收率得到18,再在盐酸作用下对18进行水解和脱羧,以92%的收率制得1。采用类似的合成策略,Bae等[40]采用氢化奎宁衍生的手性方酰胺催化115与丙二酸二甲酯的不对称共轭加成,以91%的ee值转化为114;114无需分离继续一锅法进行氢化还原和内酰胺化反应,以86%的收率制得18;最后在盐酸中水解、脱羧,以95%的收率制得1。从关键手性中间体114的光学纯度来评价,分子型手性硫脲和方酰胺催化剂催化不对称共轭加成反应的立体选择性优于固载型的手性脲催化剂,但依然存在催化剂的合成步骤冗长等问题,不利于工业化应用。
Scheme 41

Scheme 42
Scheme 43
以α,β-不饱和硝化物115为共同中间体,Bassas[41]和Baran[42]等分别报道使用奎宁丁衍生的手性硫脲和环己二胺衍生的手性方酰胺催化115与麦氏酸(Meldrum酸)进行不对称共轭加成,以75%和94%的ee值转化为116;再依次经过氢化还原和水解脱羧制得1。上述两条合成路线依旧采用手性有机小分子催化的不对称共轭加成为关键反应实现1的全合成,区别在于选用了麦氏酸替代被广泛使用的丙二酸二烷基酯作为亲核试剂。从所得手性中间体116的ee值来分析,手性方酰胺催化不对称共轭加成反应的立体选择性明显优于手性硫脲,而催化剂自身的手性骨架源对反应活性的影响不太大。
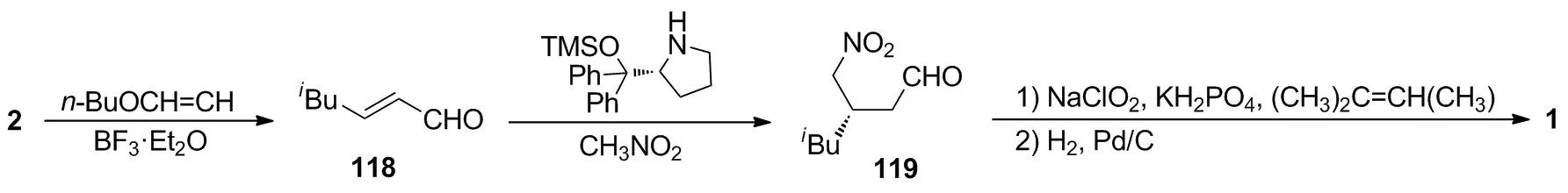
张瑜峰等[43]将2与乙烯基正丁醚在三氟化硼乙醚存在的条件下反应,以49%的收率得到α,β-不饱和醛118;然后使用手性脯氨醇催化118与硝基甲烷发生不对称Henry反应,以98%的ee值得到手性中间体119,无需纯化再依次用亚氯酸钠氧化醛基、氢化还原硝基,以3步60%的收率得到1。以α,β-不饱和醛118为共同中间体,Gotoh等[44]也报道了将118与硝基甲烷在手性脯氨醇催化作用下的不对称Henry反应,以68%收率和91%ee值得到119;119依次进行氧化、还原,以两步80%的收率制得1。两条路线的区别在于手性中间体119是否进行了分离。该路线的优势在于反应条件温和、操作步骤简单、不对称Henry反应制备的手性中间体光学纯度高。
7.6 以乙酰丙酮(120)为起始原料的不对称催化合成路线
Moccia等报道将120与羟胺进行缩合成环,产物121再进行硝化得到122,两步反应收率98%;122再与异戊醛进行缩合反应,以88%的收率得到123;将123与硝基甲烷分别在由金鸡纳啶衍生的固载型手性季铵盐非均相催化剂[45]和对应的分子型均相催化剂[46]作用下进行不对称Michael加成,以98%和90%的收率、72%和89%的ee值得到加成产物124,124的光学纯度可通过重结晶提升至99.7%;将124在强碱性反应条件下进行异恶唑的开环水解,最后进行硝基的氢化还原,以两步94%的收率制得1。以上两条合成路线的区别在于不对称Michael加成反应所采用的催化剂结构类型不同,使用固载型非均相催化剂具有反应后处理简单、便于回收再利用等工业化应用优势,但催化反应的活性中等。
7.7 以4-甲基-1-戊烯(125)为起始原料的不对称催化合成路线
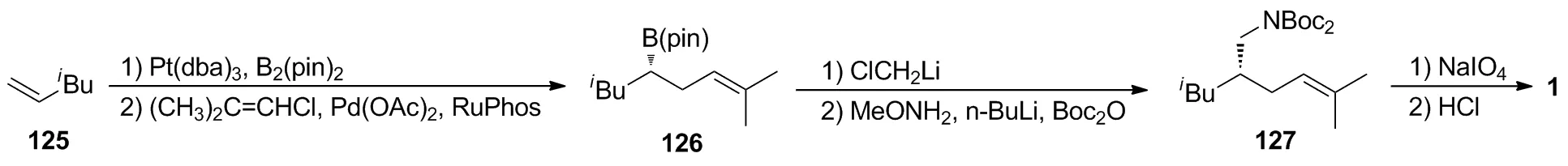
Mlynarski等[47]报道在Pt(dba)3催化作用下,将125、B2(Pin)2、手性膦配体[(R,R)-3,5-di-isopropylphenyl-TADDOLPPh]和氯代异丁烯进行一锅法串联双硼化/交叉偶联反应,以97%的收率和95%的ee值得到126;然后126在氯甲基锂、甲氧基胺和BOC酸酐作用下发生胺化和胺基保护反应得到127;127再依次被高碘酸氧化和盐酸水解脱除胺基保护,以36%的收率(以126计)制得1。该法以结构简单的单取代末端烯烃作为起始原料,通过“一锅法”对映选择性催化合成的策略,实现了高收率和高对映选择性的构建普瑞巴林C-3位手性中心。然而在上述串联反应中需要使用过渡金属铂催化以及有机膦配体的辅助作用,由于铂催化剂价格昂贵,同时有机膦化合物不易重复使用,一定程度上限制了其工业化应用。后续若能将贵金属钯和膦配体固载在无机或有机高分子材料中,使得催化剂既可以保持良好的催化活性,又能实现回收再利用,通过降低生产成本,该路线有望实现工业化应用。

Scheme 44
Scheme 45

Scheme 46
7.8 以α,β-不饱和内酰胺46为起始原料的不对称催化合成路线
Yu等[48]报道在贵金属铑与手性二烯络合物的催化作用下,将46和2-甲基丙烯基三氟硼酸钾进行不对称共轭加成,以97%的收率和99%的ee值得到128;128再依次经钯碳催化氢化还原和水解开环反应,以两步96%的收率制得1。该路线具有合成步骤少、不对称共轭加成反应收率和对映选择性高等优势,但需要使用价昂的贵金属铑催化剂,同时烯基三氟硼酸钾易产生大量有腐蚀性的副产品,不利于工业化应用。
7.9 以3-异丙基戊二腈(129)为起始原料的不对称催化合成路线
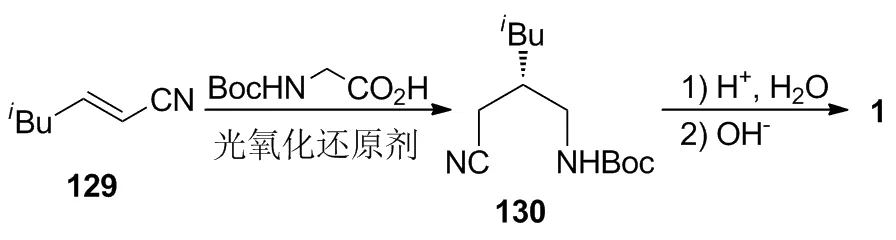
陈林等[49]报道将Boc保护的甘氨酸在光氧化还原剂和手性催化剂的催化作用下与129进行光催化不对称加成,以91%的收率得到手性中间体130;130在酸性条件下脱保护,以81%的收率制得1。该路线采用不对称光催化合成策略,仅通过两步反应实现了1的高立体选择性和高收率合成,具有节约能源、环境友好等优点,符合制药工业发展的方向。
7.10 以3-氰基-5-甲基己烯酸甲酯(131)为起始原料的不对称催化合成路线
Li等[50]报道了一条以α,β-不饱和酯131的不对称氢化还原为关键反应的全合成路线:在二茂铁-硫脲手性双磷配体和过渡金属催化剂[Rh(NBO)2BF4]的双催化反应体系中,对131进行不对称加氢还原,以96%的分离收率和98%ee值得到手性β-氰基酯132;最后对132进行选择性还原和酯基水解即可制得1。遗憾的是该路线需要使用价昂且不易回收的贵金属铑催化剂。
7.11 以(S)-3-氰基-5-甲基己酸(24)为关键手性合成中间体的不对称催化合成路线
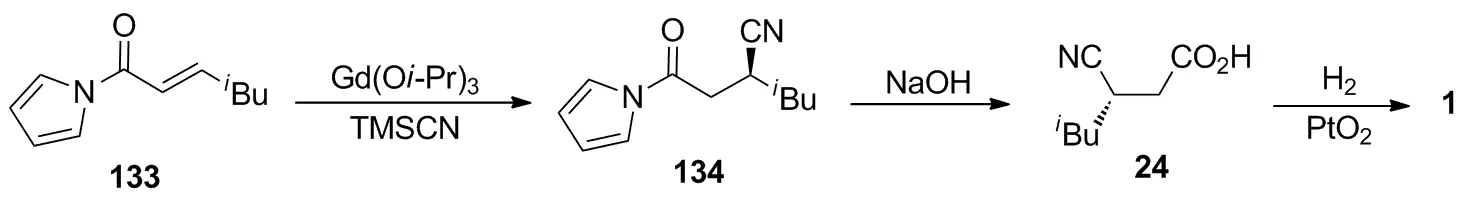
Fujimori等[51]报道了一条采用不对称催化合成策略制备手性中间体(S)-24的合成路线:在过渡金属络合物[Gd(Oi-Pr)3]和手性膦配体的催化作用下,α,β-不饱和酮133与TMSCN进行不对称氰基化反应,以99%的收率和93%的ee值得到134;先将134在氢氧化钠溶液中进行酰胺的水解,再进行氰基的氢化还原,以两步86%的收率制得1。
Sammis等[52]报道了另外一条以(S)-24为关键手性中间体的不对称催化合成路线:在金属铝和手性席夫碱络合物[(Salen)AlIII]的催化作用下,α,β-不饱和酰亚胺135与三甲基氰硅烷进行不对称氰基化反应,以97%的收率和96%的ee值生成136;然后136在氢氧化钠溶液中进行酰亚胺的水解,以94%的收率得到(S)-24;最后进行氰基的氢化还原,以92%的收率制得1。上述两条路线虽然合成步骤短,但需要使用价昂的贵金属催化剂和手性配体,不利于工业化应用。
综上所述,大量文献报道了1的全合成路线,每条路线都具有其独特的创新点,但存在不适合工业化生产的技术瓶颈或合成缺陷的路线居多。考虑到生产成本等问题,目前1的工业全合成大多仍采用外消旋体拆分的合成策略。虽然实现了拆分后另一半不需要的异构体的循环利用,但依然无法避免拆分法单步收率最高只有50%的合成弊端,导致路线的总收率偏低,且合成步骤较为繁琐。采用不对称合成的合成策略具有路线短、合成效率高等技术优势,不对称催化合成由于具有手性“增值”作用,大幅降低了生产成本,是符合绿色化学发展方向的一种不对称合成方法。然而目前1的不对称催化合成路线中大多需要使用价昂的贵金属催化剂和需要柱层析分离纯化的有机小分子催化剂,不利于工业化应用。为了降低原料药1的合成成本,为人类健康谋福祉,我们期待更多简洁高效、绿色环保的适合工业化应用的不对称合成路线问世。
猜你喜欢
分子催化(2022年1期)2022-11-02 07:10:30
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
国外医药(抗生素分册)(2016年4期)2016-07-12 14:25:19
国外医药(抗生素分册)(2016年2期)2016-07-12 14:25:01
橡胶工业(2015年2期)2015-07-29 08:29:46
西南军医(2014年5期)2014-04-25 07:42:49
食品工业科技(2014年9期)2014-03-11 18:15:39
郑州大学学报(理学版)(2014年3期)2014-03-01 04:21:06
无机化学学报(2014年12期)2014-02-28 17:34:01
