
牛病毒性腹泻病毒E2蛋白纳米抗体筛选及反应原性检测
2021-03-10 08:29李岩肖盛中杨艳张彦红蔡卓轩周子恒张哲盛金良
畜牧与兽医 2021年3期
李岩,肖盛中,杨艳,张彦红,蔡卓轩,周子恒,张哲,盛金良
(石河子大学动物科技学院,新疆 石河子 832003)
牛病毒性腹泻黏膜病(bovine viral diarrheamucosal disease,BVD-MD),是由牛病毒性腹泻病毒(bovine viral diarrhea virus,BVDV) 引起的牛等多种动物感染的一种重要传染性疾病。BVDV感染牛后,牛群出现不同程度的临床症状,可造成严重的消化系统、生殖系统、呼吸系统疾病及免疫抑制等[1-2]。该病呈世界性分布,中国、美国、阿根廷、欧洲等已普遍报道,许多养牛业发达国家均存在该病[3]。
BVDV是黄病毒科(Flaviviridae)瘟病毒属(Pestivirus)的成员[4-5],为一种单股 RNA,有囊膜的病毒,呈圆形,与猪瘟病毒和羊边界病毒为同属病毒[6]。BVDV基因组是由一个开放阅读框(open reading frame, ORF)构成的单股正链RNA[7]。根据基因组的编码和翻译,BVDV中至少包含11个成熟蛋白,包括4种结构蛋白C、Erns、E1、E2,7种非结构蛋白Npro、P7、NS2-3、NS4A、NS4B、NS5A、NS5B[8]。BVDV-E2蛋白通常可以诱导机体产生具有特异针对性的抗体,其产生的抗体往往可以中和该病毒,从而在某种程度上避免了同源毒株的攻毒和对细胞的破坏[9]。E2蛋白能够参与并介导免疫中和反应,也是病毒与宿主细胞进行识别和吸附的主要部位[10-11]。该蛋白的基因容易发生变异,因此其保守性较低,是BVDV变异较高的结构蛋白,使病毒对环境有较好的适应性[12],同时这也是导致持续性感染和疫苗保护性差的主要原因[13]。
在骆驼科动物体内,除了传统的IgG抗体以外,还存在一种天然缺失轻链和重链第一个恒定区的特殊IgG,被称作重链抗体[14]。纳米抗体的大小仅是普通IgG抗体的1/10,且结构稳定、分子量小,可以在细胞内表达,借助穿膜肽还可以透过细胞膜,具有开发成为新型抗病毒药物的潜力[15]。目前国内针对BVDV纳米抗体相关报道较少,且针对不同抗原蛋白,2015年,张国奇等[16]筛选出BVDV-NADL纳米抗体,2017年丁金花等[17]筛选出BVDV-E0蛋白纳米抗体。本试验利用BVDV灭活疫苗免疫羊驼,将抗BVDV的纳米抗体在噬菌体展示系统中展示,构建展示文库,运用E2蛋白筛选与之稳定结合的纳米抗体,为诊断和防控牛病毒性腹泻奠定基础。
1 材料与方法
1.1 材料
重组质粒pET30a-E2由南京德泰生物技术有限公司合成;BVDV灭活苗购自内蒙古金宇生物技术有限公司;羊驼一只,饲养于石河子大学兽医院;骆驼外周血淋巴细胞提取试剂盒购自Solarbio;pCANTAB5E载体、TG1和BL21均由本实验室保存;辅助噬菌体M13K07由西北农林科技大学赵钦副教授赠送;限制性内切酶NedⅠ、HindⅢ、SfiⅠ和NotⅠ以及T4 DNA连接酶均购自TaKaRa公司;DNA Marker分别购自TRANSGEN和北京天根生化科技公司;Anti-M13-HRP购自Sino Biological;Anti-E-tag-HRP购自金斯瑞生物科技股份有限公司;其他试剂均为进口或国产分析纯。
1.2 方法
1.2.1 BVDV-E2蛋白表达载体的构建及转化
预先将所需感受态细胞置于冰块上预冷,吸取10 μL连接产物加入BL21(DE3)感受态细胞,吹打混匀,置于冰块上静置30 min,预热水浴锅至42 ℃。使用计时器在42 ℃恒温水浴锅内热击90 s,立即转至冰块上静置3 min。向EP管中加入600~800 μL无抗性LB液体培养基,置于37 ℃摇床 200 r/min活化 60 min。室温条件5 500 r/min离心5 min,弃去上清,加入100 μL无抗LB培养基混匀,涂板。
1.2.2 BVDV-E2蛋白在大肠杆菌中的表达
使用接种环蘸取含有目的基因片段的菌液,在50 μg/mL的Kan+抗性LB平板上画线培养。倒置于培养箱内,37 ℃过夜。次日观察菌落生长情况,挑选单克隆,接种至4 mL Kan+(50 μg/mL)抗性的LB液体培养基中,置于恒温摇床培养至菌液OD600至0.6~0.8时,吸取1 mL菌液1 200 r/min离心1 min,弃上清后,加入80 μL去离子水和 20 μL 5×蛋白Loading Buffer,作为0 h诱导样本,-20 ℃保存。取样后向菌液中加入终浓度0.1 mmol/L IPTG,充分混匀,在37 ℃进行过夜诱导。诱导结果使用SDS-PAGE进行鉴定。
1.2.3 BVDV灭活疫苗免疫羊驼
使用BVDV灭活苗对羊驼进行免疫,分别在第0、21、49及70天采集全血,分离淋巴细胞及血清,使用分离后的血清通过ELISA检测抗体效价。取对应抗体效价较高的淋巴细胞用于后续试验。
1.2.4 引物的设计与合成
引物AIPVh-LD和CH2-R引自参考文献[18],VHH-F和VHH-R引自参考文献[19]送至北京华大基因合成。详见表 1。
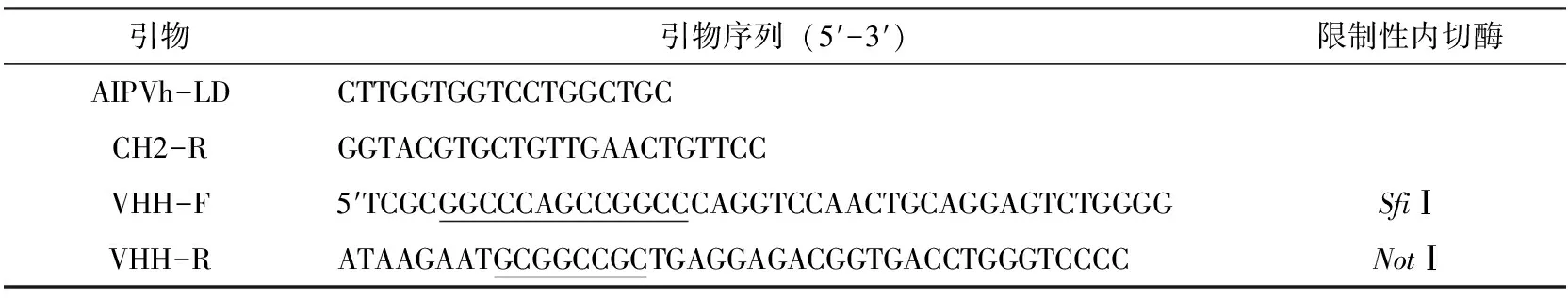
表1 目的基因扩增所需引物
1.2.5 羊驼外周血淋巴细胞RNA的提取
按照每200 μL淋巴细胞 加入1 mL TRIZol的比例 ,使用氯仿、异丙醇和无水乙醇提取总RNA,通过cDNA反转录试剂盒获得cDNA反应条件为42 ℃ 40 min,85 ℃ 5 min。
1.2.6 VHH目的片段的扩增和重组载体pCANTAB5E+VHH的构建
使用AIPVh-LD和CH2-R对获得的cDNA进行第一轮PCR扩增,反应条件为94 ℃ 5 min; 94 ℃ 30 s、57 ℃ 45 s、72 ℃ 45 s,33个循环;72 ℃ 延伸10 min,扩增后PCR产物使用1.5%的琼脂糖进行琼脂糖凝胶电泳,结束后会在900 bp和700 bp的位置出现2个条带,切取700 bp位置条带,胶回收,作为第二轮PCR模板。再用VHH-F 和VHH-R 进行第二轮PCR扩增,反应条件为94 ℃ 45 s、65 ℃ 45 s、72 ℃ 45 s,5个循环;94 ℃ 40 s、68 ℃ 45 s,35个循环;72 ℃ 延伸10 min。将扩增后的PCR产物使用1.5%的琼脂糖进行琼脂糖凝胶电泳,目的条带大约为400 bp,切取目的片段,进行胶回收,胶回收产物冻于-20 ℃保存。用SfiⅠ和NotⅠ对目的片段和质粒进行酶切并鉴定,使用T4 DNA连接酶将切好的目的片段和质粒以3∶1的比例在10 μL的体系中进行连接,反应条件为4 ℃ 16 h,连接产物放于-20 ℃保存。
1.2.7 纳米抗体噬菌体展示文库的构建
取连接好的重组质粒pCANTAB5E+VHH 10 μL 加入到100 μL TG1感受态细胞中,冰上静置30 min,42 ℃ 热激45 s,冰上静置2 min,加入900 μL AMP-LB液体培养基,放于摇床200 r/min 37 ℃摇50 min,取出后用离心机6 000 r/min离心3 min,吸出上清,再加入100 μL AMP-LB液体培养基混匀菌体沉淀,涂布到AMP-LB固体培养基上,37 ℃培养8 h。随机挑取平板中的单克隆96个,在AMP-LB液体培养基中,放于摇床200 r/min 37 ℃摇8 h后,使用第二轮PCR引物进行菌液PCR,计算库容。
1.2.8 噬菌体抗体文库救援
取构建好的噬菌体展示文库100 μL接种于100 mL 2×YT/AMP-GLU培养基中,37 ℃,200 r/min,培养至OD600值为0.6~0.8,加入100 μL辅助噬菌体(4.2×1012PFU/mL)轻轻混匀,37 ℃静置30 min,2 800g离心10 min,弃上清,菌体用200 mL 2×YT/AMP-GLU培养基重悬,37 ℃摇床培养过夜。将培养液分装至50 mL离心管,4 ℃,3 800g,离心30 min,收集上清,加入1/5体积预冷的PEG/NaCl上下颠倒混匀,冰上静置2 h。3 800g,4 ℃离心30 min后,弃上清,每管加入0.5 mL 无菌PBS 重悬菌体沉淀。12 000g4 ℃ 离心15 min 收集上清。加入1/5体积预冷的PEG/NaCl上下颠倒混匀,冰上静置2 h。10 000g4 ℃ 离心10 min弃上清,用1 mL PBS重悬噬菌体沉淀,4 ℃摇床孵育过夜,使噬菌体充分溶解。
1.2.9 重组噬菌体淘选
将E2蛋白以10 μg/孔的包被量包被到ELISA板上,另取一孔加入PBS,4 ℃包被过夜。用5%脱脂奶粉37 ℃封闭2 h。用PBST洗涤4次后37 ℃孵育稀释后的重组噬菌体2 h。用PBST洗涤5次后再用PBS洗涤3次。每孔加入0.1 mol/L三乙胺,室温静置10 min,迅速用等体积1 mol/L pH为7.4的Tris-HCl中和。收集中和后的洗脱液,并测定滴度。重复3次上述步骤,确定最后一轮淘选后噬菌体滴度。
1.2.10 重组纳米抗体的诱导表达
取第三轮筛选后洗脱产物,用2×YT稀释后取100 μL与100 μL对数生长期的TG1混合,37 ℃静置15 min。将感染后的TG1涂布于AMP/LB固体培养基上,37 ℃过夜培养。随机挑去96个单克隆菌落,依次标记,接种于100 μL LB/AMP-GLU液体培养基中,37 ℃,200 r/min过夜培养。每个单克隆取10 μL,分别接种于1 mL TB培养基中,放置于摇床37 ℃,200 r/min,培养至对数生长期,将剩余的单克隆培养样本保存于-80 ℃。菌液OD600到达0.6~0.8时每管菌液加入含10 mmol/L IPTG的TB培养基中,放于摇床37 ℃,200 r/min诱导过夜。将过夜诱导产物4 ℃ 3 200g离心10 min,弃上清,置于-20 ℃冷冻40 min,取出室温放置15 min,每管加入无菌PBS 500 μL,重悬菌体,放于摇床200 r/min 30 min。取出后放于离心机,4 ℃ 3 800g离心15 min,收集上清(上清即为可溶性纳米抗体粗提物)。
1.2.11 可溶性重组纳米抗体的ELISA检测
将E2以10 μg/mL的包被量包被在ELISA板上,同时包被两板,4 ℃过夜,空白孔直接加入PBS作为无抗原对照。弃去孔中包被液,拍干后加入200 μL 5%脱脂奶粉,37 ℃封闭2 h。用PBST洗板3次,取可溶性纳米抗体,用封闭液1∶1稀释后,每孔100 μL,37 ℃孵育40 min。用PBST洗板3次,两板分别孵育稀释好的抗M13抗体和抗E-tag抗体,37 ℃孵育40 min。用PBST洗板3次,加入显色液,37 ℃显色15 min,每孔加入50 μL 2 mol/L硫酸终止,测量OD450的值。
2 结果与分析
2.1 pET30a-E2重组质粒酶切鉴定
使用NdeⅠ和HindⅢ对构建pET-30a-E2原核表达载体进行双酶切验证。结果如图1,双酶切后,E2目的基因片段与预期分子量相符。
2.2 E2蛋白诱导表达
表达菌在37 ℃经0.1 mmol/L IPTG诱导后,经SDS-PAGE鉴定,如图2所示,在39 kDa左右出现与预期大小相符的条带,表明蛋白均成功表达。
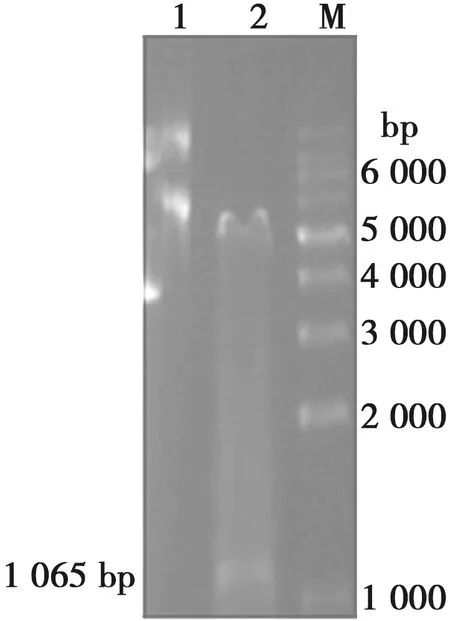
1. 质粒对照;2. 双酶切产物;M. DNA Marker
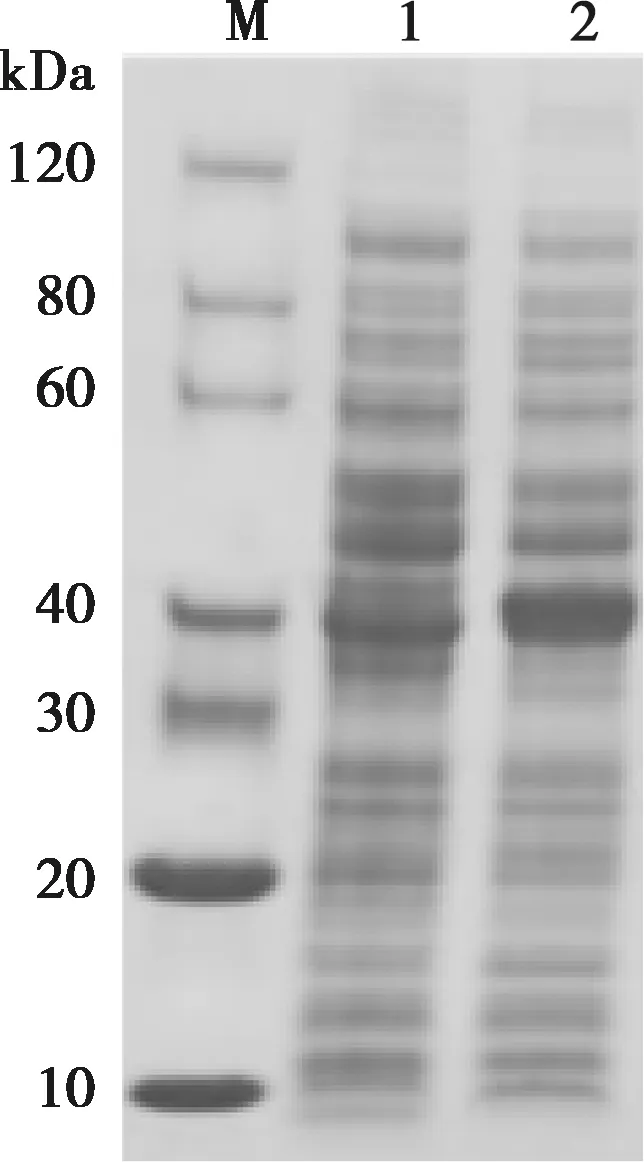
M. 蛋白Marker;1. 0 h对照;2. 37 ℃诱导16 h
2.3 VHH目的片段扩增结果
经过两轮PCR扩增,产物经琼脂糖凝胶电泳后结果如图3,在400 bp左右出现目的条带。
2.4 重组质粒pCANTAB5E+VHH的酶切鉴定
将连接好的pCANTAB5E+VHH用限制性内切酶SfiⅠ和NotⅠ进行酶切鉴定,在4 500 bp左右和400 bp左右出现2个条带,结果如图4。
2.5 文库容量鉴定
通过随机挑取的96个单克隆的菌液PCR琼脂糖凝胶电泳结果,计算库容。结果如图5、图6。阳性率为90.8%,库容为4.9×1014CFU/mL。
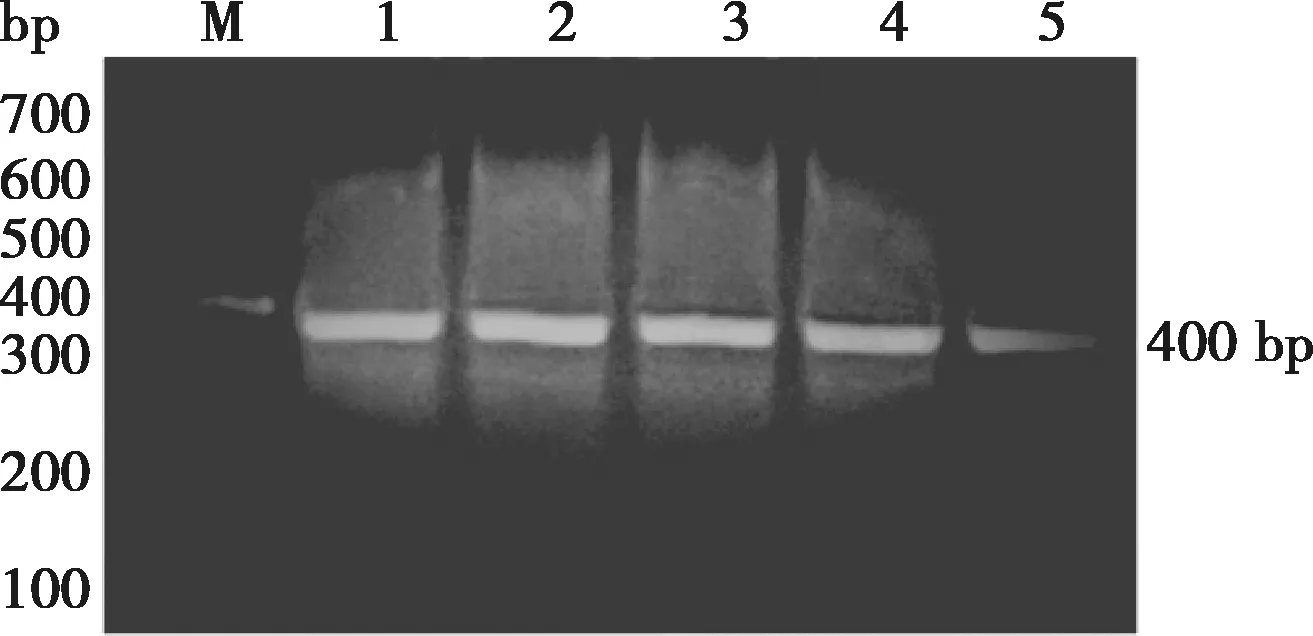
M. DNA Marker;1~5. PCR产物
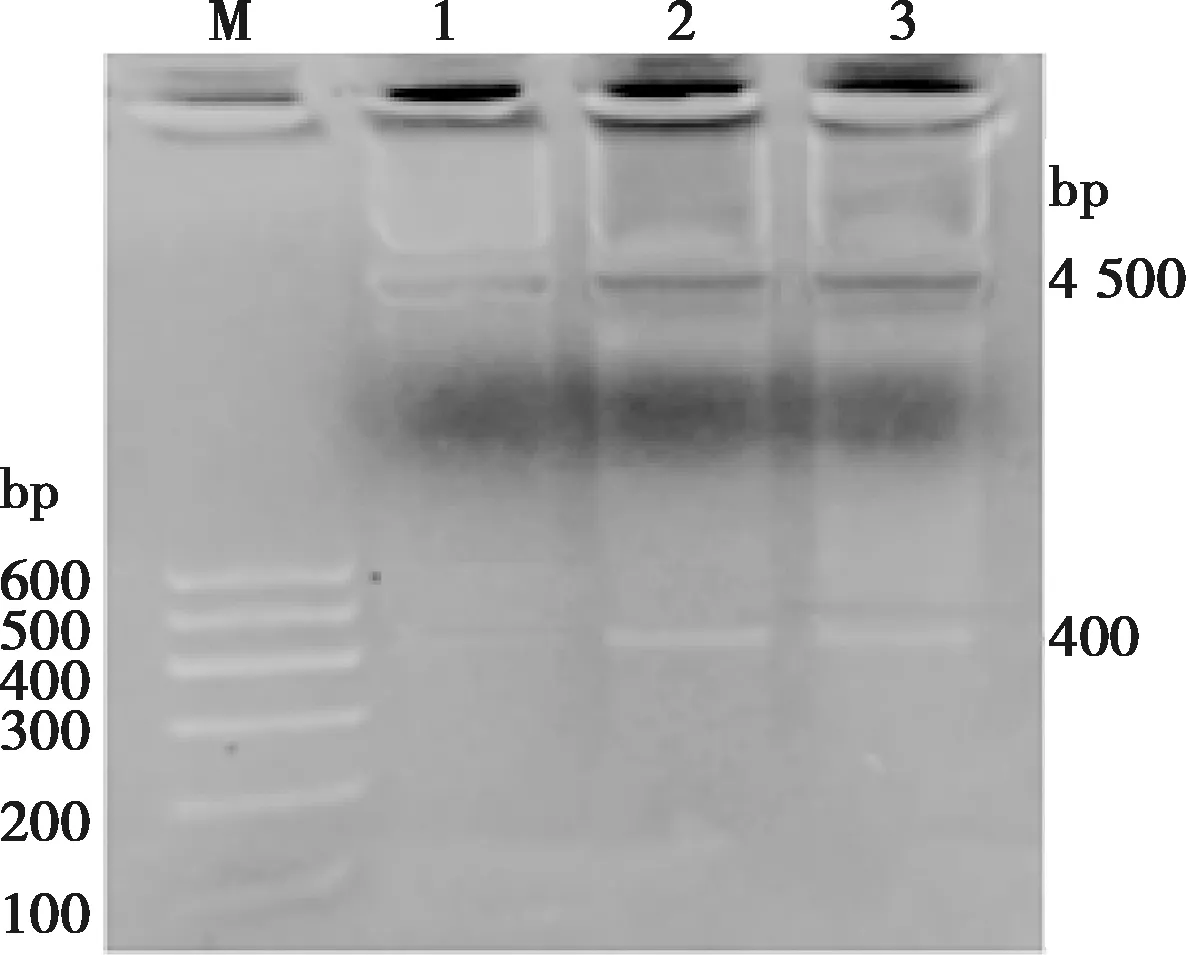
M. DNA Marker;1~3. pCANTAB5E+VHH酶切产物
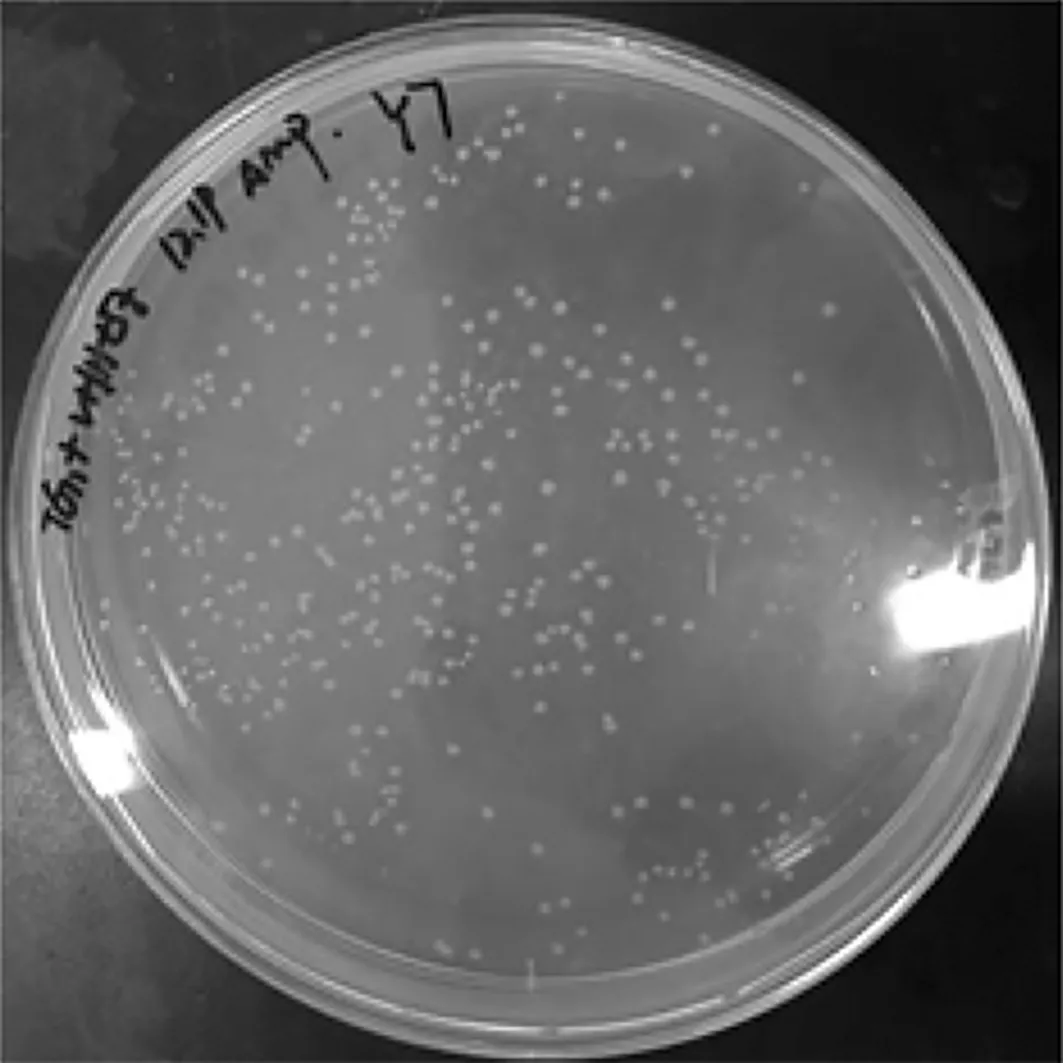
图5 文库库容量鉴定
2.6 重组噬菌体淘选
将E2蛋白作为包被抗原筛选特异性纳米抗体。在筛选过程中,通过检测每轮洗脱液中重组噬菌体的滴度来评估特异性VHH 重组噬菌体的富集效果。结果如表2。根据表2,噬菌体的加入量记为Input,洗脱量记为P,阴性对照孔中噬菌体洗脱量记为N,P/Input表示回收率,P/N值表示特异性噬菌体所占比。结果显示特异性噬菌体得到了明显的富集。
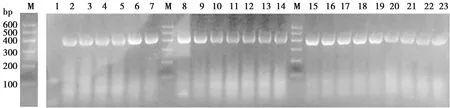
图6 菌液PCR鉴定VHH文库阳性率
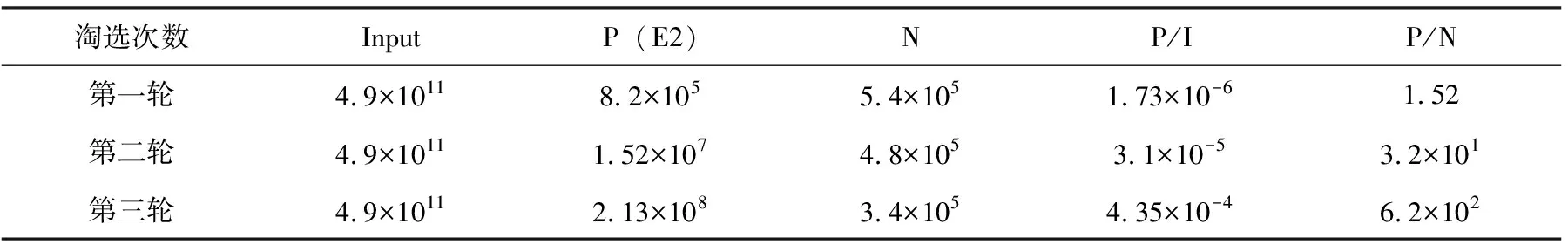
表2 E2蛋白筛选过程中特异性噬菌体富集情况
2.7 Phage ELISA
从E2蛋白第三轮筛选后的细菌平板上随机挑取96个单克隆,经扩增诱导后,制备可溶性重组纳米抗体粗提物,分别用HRP鼠抗M13和HRP标记的E-tag作为二抗,通过 ELISA检测,初步鉴定纳米抗体克隆与E2重组蛋白的反应性,ELISA结果如图7、图8所示。图中横坐标为单克隆菌落序号,纵坐标为OD600的值,以OD600的值超过对照组4倍为阳性样本,进行后续试验。
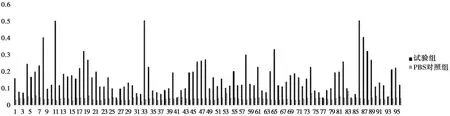
图7 ELISA检测重组纳米抗体与E2的反应性(HRP鼠抗M13)

图8 ELISA检测重组纳米抗体与E2的反应性(E-Tag)
选取两组ELISA均为阳性,且OD值较高的阳性克隆进行菌液PCR,将PCR结果送至北京华大基因测序,测序结果经序列分析后,得到与驼源的VHH具有较高同源性且与E2蛋白具有良好反应性的序列2条。分别命名为Nb-YT1、Nb-YT2。其中在其FR2区均由典型亲水氨基酸,且在其CDR3区均有半胱氨酸,根据文献报道,一对半胱氨酸可形成二硫键,稳定纳米抗体抗原结合区的结构。氨基酸序列如图9。

图9 特异性纳米抗体氨基酸序列
3 讨论
有研究表明,BVDV的结构蛋白变异性较大,特别是E2的变异种类最多[20]。变异的结果通常会导致BVDV对宿主细胞以及环境的适应能力更好,生存能力更强,同时这也是导致某些疫苗以及药物的功能性丧失的重要原因[21]。由于E2能诱导机体产生中和抗体,故目前研制的新型疫苗多为针对E2蛋白。E2基因在 BVDV不同毒株以及与瘟病毒属各毒株之间高度不稳定[22]。E2的高度不稳定性使 BVDV更好的适应环境,这也是导致疫苗保护失效和持续性感染的主要原因[23]。本研究成功获得了BVDV-E2纳米抗体氨基酸序列,纳米抗体不但可以用于研究E2蛋白的表达和功能,还可以用于为BVDV的检测与预防。
纳米抗体具有特异性识别位于病毒表面、病毒缝隙甚至被包埋的病毒抗原表位的功能,因此,纳米抗体具有优越于传统抗体的结合能力,从而高效率地并高针对性地靶向目标病毒,实现中和病毒的功效[21]。2015年张国奇[16]筛选出来针对BVDV-NADL株的纳米抗体对病毒的复制有阻断效果;2017年丁金花[17]筛选出来针对BVDV-E0的纳米抗体均能与病毒发生特异性反应,细胞试验结果表明其具有细胞毒性。
本试验通过全病毒免疫羊驼,分离全血中的淋巴细胞,提取总RNA,通过两轮PCR扩增,得到目的片段,将其与表达载体连接后,通过噬菌体展示技术,获得噬菌体展示文库。本试验获得的文库库容为1.02×107CFU/mL,插入率为90.8%。经过3次淘选,挑取第三轮的洗脱产物与对数生长期TG1结合后的单克隆,使用IPTG进行诱导表达。表达产物通过ELISA测试反应性,OD值较高的克隆测序结果显示有2个与驼源VHH具有较高同源性且不完全一致。本试验获得的Nb-YT1、Nb-YT2的氨基酸序列可通过大量表达纯化后,进行标记,用于BVDV致病机理、诊断及治疗。
猜你喜欢
昆明医科大学学报(2022年2期)2022-03-29
中国动物传染病学报(2021年3期)2021-07-21
煤气与热力(2021年4期)2021-06-09
中国现代医药杂志(2020年10期)2020-12-14
中国茶叶(2017年1期)2018-01-04
现代检验医学杂志(2016年3期)2016-11-15
广东海洋大学学报(2015年3期)2015-12-22
医学研究杂志(2015年3期)2015-06-10
特产研究(2015年1期)2015-04-12
沈阳医学院学报(2014年4期)2014-12-27
