
非典型的遗传性弥漫性白质脑病合并轴索球样变综合征1例报告并文献复习
2021-03-09 11:10汪国宏
中风与神经疾病杂志 2021年12期
汪国宏, 潘 静
遗传性弥漫性白质脑病合并轴索球样变(leukoencephalopathy,hereditary diffuse,with spheroids,HDLS),虽存在散发病例报告,在全球来说仍为罕见的常染色体显性遗传病。HDLS临床症状多样且具有异质性,个体间差异显著,即使不同个体存在同一家系内相同突变的基因,而个体之间临床表现仍复杂多变。同时此类疾病显著的MRI影像表现存在非特异性,从而导致HDLS的临床诊断率较低。此文报道1例临床初步诊断为脑小血管疾病,最终基因检测明确为临床意义未明变异的HDLS疾病。我们将回顾性分析HDLS临床表现、基因、脑成像等辅助检查及遗传特征,从而复习总结相关文献提高临床辨别率。
1 病例资料
患者,女性,47岁,大专文化,主因“进行性记忆力减退2 y,加重伴行动迟缓、左下肢拖拽7 m”于2020年11月16日入院。1 y前(2019年12月)无明显原因下出现头晕,记忆减退,就诊于当地某医院,头部MRI检查显示:侧脑室旁多发腔隙灶,DWI及FLAIR示点状高信号,急性脑梗死。脑电图:未见异常。按“脑血管病”收住治疗,症状无明显改善。2020年6月患者出现记忆减退逐渐加重、缺乏主动性、排斥工作等情绪认知障碍及动作迟缓,再次就诊当地医院仍考虑“脑小血管病”。近7个月来逐渐出现行走拖曳、行动迟缓等症状进一步加重,遂入住上海瑞金医院神经内科。既往史:体健。家族史:否认家族遗传史,育一女,体健。神经系统查体:反应迟钝,空间定向力、时间定向力、记忆力、计算力粗测尚可;双侧肢体腱反射(),四肢肌力均为5级,左侧肢体肌张力增高,右侧肢体肌张力正常;双侧轮替、拍指运动欠灵活,左侧重于右侧,左手失用;脑膜刺激征(-);左侧巴宾斯基征(+),余病理征(-);左下肢拖曳,行走时左侧肢体摆臂减少。辅助检查:血常规、动态红细胞沉降率、生化全项+同型半胱氨酸、糖化血红蛋白、维生素B12+叶酸、甲状腺功能全项、肿瘤全项、风湿3项+免疫5项、自身免疫性抗体等均未见异常。神经心理学检查:使用简易精神状态检查(Mini-Mental State Examination,MMSE)和蒙特利尔认知评估量表(Montreal Cognitive Assessment,MoCA)评估患者整体认知功能。影像学检查:使用头部MRI进行脑部一般情况检查。使用18F氟代脱氧葡萄糖(2-deoxy-2-fluoro-glucose,FDG)正电子发射断层摄影术(positron emission tomography,PET)检查以评价患者脑葡萄糖代谢。脑电图检查及基因检测:使用DYD2000数字视频脑电图仪,按国际10-20系统安放盘状电极,进行描记脑电图,记录时间2 h。对患者进一步采用基因测序检查。神经心理学评估量表测定结果:粗略测评患者MMSE评分为27分,MoCA评分为18分。影像学检查:头部MRI示双侧丘脑、侧脑室体部、前后角旁及放射冠区广泛白质病变;脑萎缩(见图1)。头部PET/CT:双侧侧脑室前角周围额叶及后角周围顶枕叶脑白质密度弥漫性下降,后角处较前角处严重,右侧较左侧严重,提示脑白质病变(见图2)。脑电图检查及基因结果:脑电图检查示未见明显异常。临床表型相关基因变异:CSF1R 基因上1个杂合错义变异c.2473G>C:p.E825Q(见图3)。
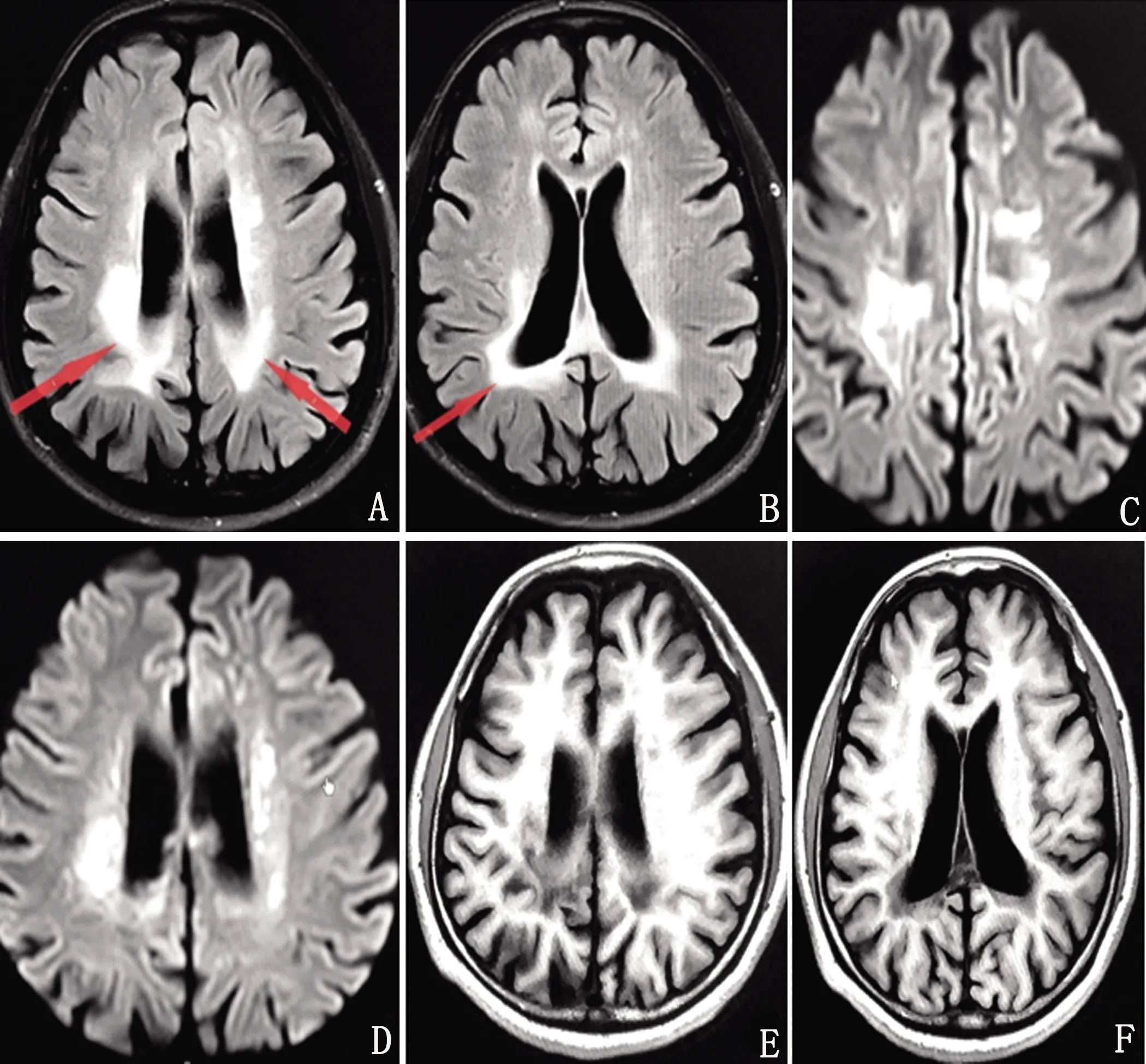
图A、B:头部MRI平扫示双侧丘脑、侧脑室体部、前后角旁及放射冠区可见大片片状异常信号灶,FLAIR呈稍高信号;图C、D:DWI呈明显高信号灶;图E、F:T1W呈稍低信号

图A~F:头部 PET/CT检查可见双侧基底节区见钙化灶;双侧侧脑室前角周围额叶及后角周围顶枕叶脑白质密度弥散性下降,后脚处较前角处严重,右侧较左侧严重
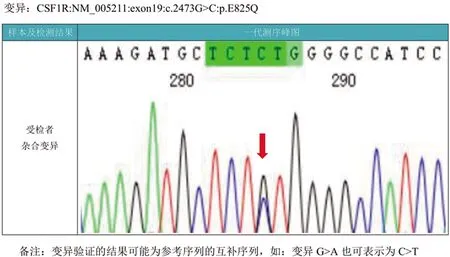
图3 患者一代基因测序图。箭头处为突变位点c.2473G>C:p.E825Q
2 讨 论
HDLS是一种非常罕见的家族性进行性神经退行性疾病,主要影响大脑白质。Axelsson在1984年[1]等首次报道该病并命名,迄今已报道100多例HDLS患者。该病发病常见年龄在20~80岁之间,较多于40岁左右起病,病程可长达11 y,平均病程约6 y。此病临床表现为主要有两组症状:神经精神症状和运动症状。其最具代表性的临床表现主要有高级皮质功能的损害(如记忆力下降、执行功能减退、行为甚至人格改变)、运动和感觉障碍、帕金森症状和肌张力障碍、癫痫样发作、额叶症状(如缺乏自制力、判断力丧失、洞察力下降等)、小脑及脑干症状。最终HDLS患者大多数因为肌强直、肌痉挛而长期卧床,逐步失语,直至植物人状态。就现有病例总结HDLS患者多见是由于肺部感染及其他继发性感染而死亡。HDLS显著的MRI影像表现[2~4]为早期T2/Flair高信号,T1低信号,表现为双侧、非对称性及局限性,以额叶、顶叶及枕叶为著,主要累及皮质深部以及皮质下区、脑室周围白质束。其中胼胝体异常信号及发育不良(被认为是疾病的早期特征),也可见到弥漫性脑组织萎缩、脑室扩大以及皮质脊髓束受累[2,5,6]。处于病情后期,可观测到病灶相互融合后对称性片状分布。额叶白质的早期变化可能是HDLS患者主要的认知障碍和精神症状的原因。此外HDLS显著的病理学特征[7~9]是光镜下可见胶质细胞着色,神经轴索球样变以及弥漫性轴索变性,髓鞘缺失。
2011年,Rademakers等[7]运用全基因组连锁分析和外显子测序确定了该病基因变异位点位于5q32的集落刺激因子1受体(colony-stimulating factor-1 receptor,CSF1R),这是一种细胞表面受体,在发育和先天免疫中起关键作用,不同的基因突变可能与不同的临床表型有关。CSF1R基因是目前唯一被确定为HDLS的致病基因,是调控巨噬细胞增殖、分化和功能的细胞因子,从属于CSF1/PDGF受体酪氨酸激酶家族。CSF1R基因存在70种突变(错义、大片段缺失、移码、无义及复杂重排等)。酪氨酸激酶结构域(12-22外显子)是常见的突变地点,外显子18-20为突变的高发区。CSF1R是小胶质细胞发育和维持的重要因素,因CSF1R激酶结构域的突变,进而引起小胶质细胞增殖分化及表达受阻,最终致脑内小胶质细胞功能发生损害,而小胶质细胞功能障碍在HDLS发病机制中具有重要作用[10~12],但目前具体致病机制仍不明确。另外有学者利用全外显子组测序在CSF1R基因的内含子16 中发现了一个新的剪接位点突变(c.2319+1C>A),受影响患者的外周血样本中CSF1R mRNA显著降低。研究结果不仅支持这种剪接位点突变的病理意义,还首次证明了对CSF1R表达的显性负效应[13]。
另外,研究发现CSF1R亦是色素型正染性脑白质营养不良(pigmentary orthochromatic leukodystrophy,POLD)的致病基因,因HDLS和POLD的临床表型和神经病理学特征有重叠,且均可检测到CSF1R突变,现将其合并统一称为“成人起病的脑白质病伴轴索球样变和色素胶质细胞” (adult-onset leukoencephalopathy with axonal spheroids and pigmented glia,ALSP)[8]。而有研究发现在病理证实的HDLS病例中并不总能发现CSF1R突变。由于HDLS表型多种多样,且部分存在非典型的表现,加上对该病认识不足,该病的发病率可能被远远低估,临床及易被误诊为痴呆、癫痫、多发性硬化、帕金森病或综合征(多系统萎缩、皮质基底节变性)、脑卒中、脑白质营养不良等疾病[14]。根据多中心研究及回顾性分析,2017年提出特异性较高(>96%)的参考诊断标准[15]。于临床中,不仅根据临床症状的变化(如精神人格改变、渐进性记忆力、认知功能的减退等),询问是否存在相关的阳性家族史,以及辅助检查中典型脑白质影像学改变,在与多种其他相关疾病鉴别之后,可考虑为HDLS,但最终需要结合基因检测CSF1R突变进行确诊。
根据诊断标准,我们报道此次病例满足核心特征中记忆认知衰退(空间定向力、时间定向力、计算力、近期记忆力减退)、性格行为改变及精神症状(缺乏主动性、排斥情绪)、运动步态障碍(左侧偏瘫步态)、非对称帕金森综合征(行动迟缓)、锥体束受累(病理征阳性)、高级皮质受损(左手失用)、头部MRI提示双侧侧脑室周围出现典型的脑白质病变,以及患者所检测基因型CSF1R:NM_005211:exon19:c.2473G>C:p.E825Q,参照美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics,ACMG)相关指南[16],该变异为临床意义未明变异。
目前已有超过90多种致病性CSF1R突变报道,但还没有明确的表型-基因型相关联性[17]。大多为错义突变,移码,无义突变,插入/缺失,以及剪切位点的突变也都有病例报道[5,7,8]。此次发现受检者CSF1R 基因第825位密码子由编码谷氨酸变为编码谷氨酰胺,检测主因为CSF1R基因存在1个杂合错义变异c.2473G>C:p.E825Q。虽然目前该患者变异尚无文献报道,但结合其临床表现、辅助检查等,可明确诊断为HDLS。
针对HDLS治疗,现并未寻之切实有效的针对性治疗,临床上多以对症治疗为主,其中包括加强护理,抗痫、抗生素的应用,营养支持综合治疗。也有学者正在研究通过条件遗传方法或药物疗法(CSF1R 抑制剂)替代小胶质细胞耗竭后的空小胶质细胞生态位的免疫疗法来治疗[18]。国内有报道经异基因造血干细胞移植治疗的1例HDLS患者,其病情得到有效控制和延缓[19]。一般该病基因型和表型无相关,但该病表型多样性,基因变异型亦在不断丰富、被发现,本例即是一种全新的基因突变临床意义未明,今后在CSF1R突变是怎样影响基因表达、信号传导,以及相关异常代谢通路在白质脑病及轴索和髓鞘病变中等方面的详尽机制,都还有待于进一步探究,为今后该病的诊治方法带来更多依据。
猜你喜欢
锦州医科大学报(2022年3期)2022-06-06
东南大学学报(医学版)(2021年6期)2022-01-27
影像研究与医学应用(2021年15期)2021-09-12
昆明医科大学学报(2021年3期)2021-07-22
昆明医科大学学报(2021年3期)2021-07-22
神经损伤与功能重建(2020年10期)2020-12-23
神经损伤与功能重建(2020年11期)2020-12-01
中国CT和MRI杂志(2020年2期)2020-03-10
医学新知(2019年4期)2020-01-02
保健与生活(2019年15期)2019-09-12
