
非同义单核苷酸变异致病性预测研究综述
2021-03-09 02:34:30朱一亨於东军
南京理工大学学报 2021年1期
葛 芳,胡 俊,朱一亨,於东军
(1.南京理工大学 计算机科学与工程学院,江苏 南京 210094;2.数据科学与智能应用福建省高校重点实验室,福建 漳州363000;3.浙江工业大学 信息工程学院,浙江 杭州 310023)
在生命体内,遗传变异(如基因型或表型变异)可能会导致蛋白质结构的改变,对蛋白质的稳定性、功能产生影响[1,2],从而导致疾病的发生,如骨髓增生性疾病、肾上腺肿瘤、脑癌等[2,3]。由此可见,遗传变异与人类身体健康关系密切。随着生物技术进步,全基因组/全外显子组测序成本降低,从人类基因组中鉴定出的遗传变异数量也在剧增。通常,这些鉴定出的变异分为5类,分别是染色体非整倍性(Chromosomal aneuploidy,CA)[4]、结构变异(Structural variations,SV)[5]、复制数变异(Copy number variations,CNVs)[6]、短插入/删除(Short insertion/deletions,indels)[7]和单核苷酸变异(Single nucleotide variations,SNVs)[8]。发生在编码区的SNVs,又被划分为同义(Synonymous)和非同义(Non-synonymous)两种类别[9]。在上述人类遗传变异的研究中,突变和变异是两个常见的概念。相关学者把全球人类群体作为一个整体,在小于 1%的人群中检测到的变异,称为突变(Mutation);反之,大于1%的变异称为多态性(Polymorphism);变异(Variations)是突变和多态性的统称[10]。多态通常表现为人类身体外观的改变,如肤色、身高、眼睛等,并不具备致病性;而突变极有可能引起人类疾病,影响人类生存[10,11]。
在所有的人类遗传变异类型中,非同义单核苷酸变异(Non-synonymous single nucleotide variations,nsSNVs)约占90%[12]。已有研究表明,近三分之一的nsSNVs对人体健康有害,可导致疾病[3]。据记载,超过6 000种的人类疾病是由该类变异引起的,例如:囊胞性纤维症、马方综合征、早老性痴呆、癌症等[11]。除了对人体有害的nsSNVs外,还有约三分之二的nsSNVs并没有改变蛋白质的结构和功能表达,对人体并不构成危害,表现为中性。如前所述,有害的nsSNVs可导致一般性疾病,甚至可导致癌症。针对癌症而言,在癌症基因组中检测到的大部分氨基酸替换对癌症产生和发展的影响很小或者没有影响,此类nsSNVs称为乘客突变(Passenger mutation),仅有小一部分nsSNVs会导致癌症,被称为驱动突变(Driver mutation),如血癌、口腔癌等[13-15]。准确地区分驱动突变和乘客突变,对癌症的早期发现和治疗有着重大意义。
下一代测序技术的最新进展为人类基因组分析提供了大量的突变数据,及时有效地鉴别出与疾病相关的突变显得十分有必要[11]。在传统生物医学上,临床医生往往通过分析患者的基因测序数据,判断该患者的病症是否为nsSNVs所引起。由于基因测序数据的记录不完整、未考虑家族病史、对数据分析不够全面等,临床医生往往不能快速准确地判断检测出的变异是否具有致病性,从而给治疗方案的设计带来不便[16]。同时,传统生物医学手段很难发现尚未引发疾病的致病性nsSNVs,无法指导人们做好预防工作。此外,利用传统生物医学手段来判断nsSNVs是否具有致病性的周期长、代价高。为了解决上述问题,相关研究学者利用生物计算技术来预测nsSNVs致病性,提出了多种快速准确的预测方法[17-19],这些方法有助于了解遗传疾病的发病机制,对制定疾病预防策略及对应新药物设计等有着至关重要的意义[20-22]。
1 非同义单核苷酸变异
一个人基因组中大约有300万个SNVs,约占人体基因组所有碱基的千分之一[23]。这种变异可能发生在基因间区、启动子区、编码区、UTR区(Untranslated region),或者某些非编码核糖核酸(Ribonucleic acid,RNA)区[24]发生在编码区的SNVs有同义突变(Synonymous mutation)和非同义突变(Non-synonymous mutation)两种类别。非同义突变有无义突变(Nonsense mutation)、错义突变(Missense mutation)和移码突变(Frameshift mutation)3种类别。发生在非编码区的突变和编码区的同义突变又称为沉默突变(Silent mutation),该类型突变不会改变蛋白质翻译。在所有遗传变异类型中,错义突变是人类遗传变异中最常见的一类,根据错义突变是否对人体产生危害,分为中性和非中性突变(即致病性突变)两种类别[9]。图1给出了单核苷酸变异的类型。
发生在编码区的SNVs分为同义、无义、错义和移码突变4种类别,如图2所示。
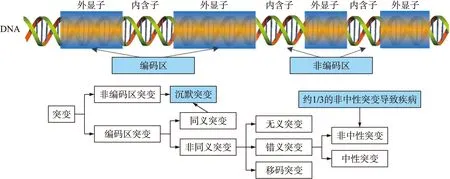
图1 单核苷酸变异类型
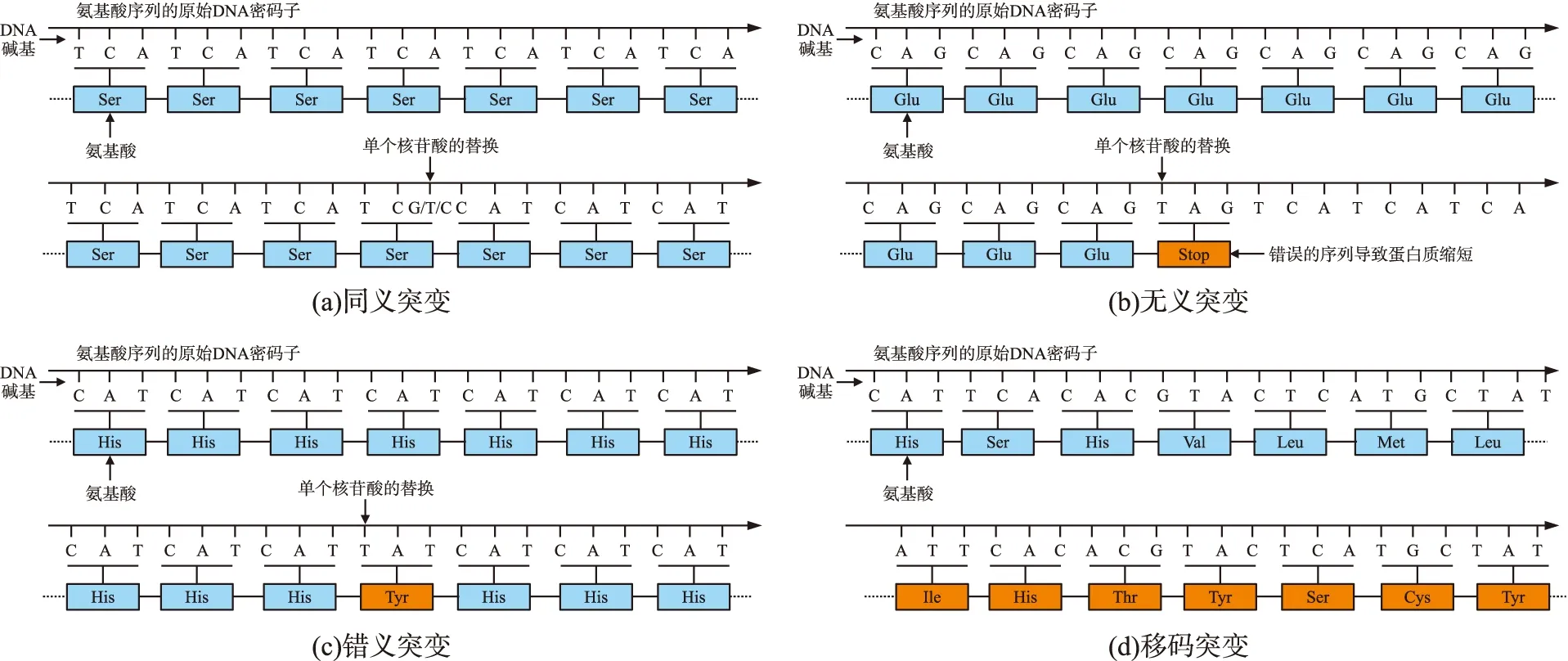
图2 编码区的SNVs
(1)同义突变指编码后的蛋白质不发生改变。如图2(a)所示,在编码亲水性丝氨酸(Serine,Ser)的密码子TCA中,腺嘌呤(A)突变为鸟嘌呤(G)/胸腺嘧啶(T)/胞嘧啶(C),密码子相应突变为TCG/TCT/TCC。由遗传密码矩阵可知,突变后的3种密码子仍编码为丝氨酸;
(2)无义突变又称为终止获得型突变,突变导致蛋白质翻译过程提前终止。如图2(b)所示,在编码谷氨酰胺(Glutamine,Glu)的密码子CAG中,胞嘧啶(C)突变为胸腺嘧啶(T),突变后的密码子为TAG。由于TAG是一种终止密码子,所以,突变导致蛋白质翻译过程提前终止;
(3)错义突变是一种最常见的SNVs,突变后,编码的蛋白质发生改变。如图2(c)所示,在编码碱性组氨酸(Histidine,His)的密码子CAT中,胞嘧啶突变为胸腺嘧啶,突变后的密码子为TAT,编码的氨基酸也相应突变为疏水性的酪氨酸(Tyrosine,Tyr);
(4)移码突变是指单核苷酸的插入或删除,导致密码子移位。如图2(d)所示,删除密码子CAT中的胞嘧啶(C),导致密码子均产生一个移位,使得编码的蛋白质均发生改变。
2 研究现状
近些年,随着生物测序技术的发展,大量的nsSNVs数据库为该领域研究人员提供了基础数据来源[25-27],基于生物计算的技术被应用到nsSNVs致病性预测问题,并且涌现了大量相关研究工作[17-19]。根据使用特征的不同,可分为6种类别,分别是:(1)基于突变频率的方法,如Mutation significance(MutSig)[28];(2)基于通路的方法,如HotNet2[29];(3)结合基因组和转录信息的方法,如Combined annotation dependent depletion(CADD)[30];(4)基于序列进化保守性的方法,如Sorting intolerant from tolerant(SIFT)[31];(5)基于序列和结构混合特征的方法,如Polymorphism phenotyping v2(PolyPhen-2)[32];(6)综合评价类方法,如Consensus deleteriousness score(Condel)[33]。表1给出了6类nsSNVs致病性研究的代表性方法和对应网址。
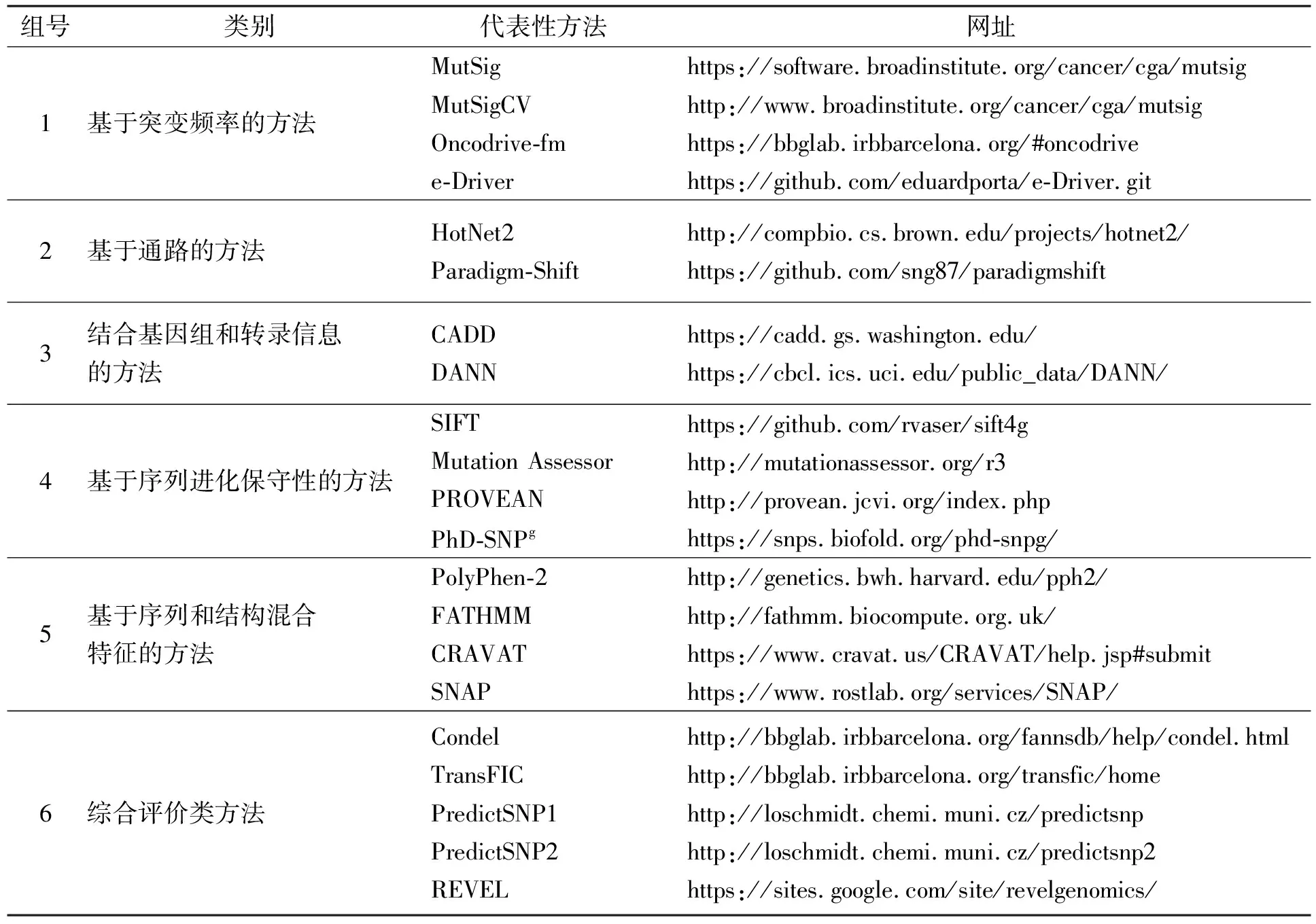
表1 6类nsSNVs致病性研究的代表性方法和对应网址
2.1 基于突变频率的方法
基于频率的突变预测方法是早期癌症驱动基因分析的常用方法,主要通过观测突变频率在中性样本(非致病性突变样本)和疾病样本(致病性突变样本)中的差异来进行判断,最具代表性的有MutSig[28]和Oncodrive-fm[34]。
MutSig[28]通过分析基于背景突变率(Background mutation rate,BMR)的相对突变丰度、基因内热点突变聚类情况,以及突变是否发生在被保护区域等3个方面,来预测突变的危害性。MutSig[28]的改进版Mutation significance covariates(MutSigCV)[35],也将上述3个方面作为突变致病性判断的核心因素。另外,MutSigCV将“聚类”和“保护”联合计算,实现在保守热点区域内聚类,避免突变特性的重复计算[35]。在计算BMR时,MutSigCV发现邻居(Neighbor)基因与中心基因具有相似的基因组特性,如脱氧核糖核酸(Deoxyribonucleic acid,DNA)复制时间、染色质状态(开放/封闭)以及转录活性的一般水平(高转录与未转录)等;此外还发现,类似基因组参数与背景突变率紧密相关,因此,MutSigCV将此类协变量纳入背景模型,通过汇集协变量空间的邻居基因数据来改善BMR估计,实验结果证明,MutSigCV可明显减少假阳性(False positive)数量[35]。
由于MutSig[28]和MutSigCV难以鉴别具有低复发频率的驱动基因[11],Abel等人提出了Oncodrive-fm[34]方法,这是一种不依赖于突变复发的候选癌症驱动基因检测方法。该方法检测多个肿瘤样本,发现具有高功能影响(Functional impact,FI)趋势的基因或模块,与此同时,重点评估2个指标:(1)跨多个肿瘤样本,评估所有基因的体细胞SNVs的FI值;(2)评估每个基因或基因模块中FI变异的重要性[34]。因此,在检测候选驱动基因或基因模块时,Oncodrive-fm可以有效识别低复发性候选癌症驱动因子。
在识别癌症驱动基因中,MutSig[28]和Oncodrive-fm[34]均将基因作为基本单元,并假设基因是癌症中具有特定作用的实体,Porta-Pardo等人认为针对不同区域的突变,对整个基因的影响可能并不相同,与癌症的相关性也可能不尽相同;同一蛋白质内不同功能区域(Protein functional regions,PFR)在致癌机制中的作用也可能不同。针对上述问题,Porta-Pardo等人开发了e-Driver[36]。该方法通过识别一个蛋白质不同PFR的体细胞错义突变,发现该蛋白质中具有突变偏移的区域,包括域(Domains)或内部无序区域(Intrinsically disordered regions,IDRs),解释了具有突变偏移的蛋白质可能是真正的癌症驱动力[36]。
2.2 基于通路的方法
基于细胞信号传导与调节通路的预测方法,在预测突变是否对人体健康产生危害时,通常会考虑突变的生物效应,即基因间的相互作用和已知的生物通路,代表性方法有Paradigm-Shift[37]和HotNet2[29]。
Sam等人指出,若焦点基因(Focus gene,FG)突变后导致功能丧失(Loss of function,LF),理论上会在转录后的表达水平有所体现,即当检查FG下游基因的活动时,可能会发现FG不活跃的证据。类似地,若FG发生功能获得型突变(Gain of function mutation,GFM),理论上也会在调控通路的上下游有所表达[37]。基于该理论背景,Sam等人于2012年提出了Paradigm-Shift方法[37]。在一组通路相互作用的背景中,该方法使用置信传播算法推断基因表达活性和拷贝数情况,同时检测其下游邻域中基因的预期活性相对于其上游的预期差异,利用基因的已知遗传相互作用,激活或失活这些相互作用的基因,从而判定突变是否导致蛋白质功能丧失或获得。该算法使用来自基因下游输出的预测结果调节上游输入,因此被认为能够为突变注释提供更准确的信息[37]。
Leiserson等人于2015年提出的HotNet2[29],使用“绝缘”热扩散理论,对扩散网络中的热量来源进行编码,分析基因突变及其局部拓扑结构,最终发现具有较高热分数的“热”子网,鉴别出具有显著突变相互作用的基因组。具体地,HotNet2对来自癌症基因组图谱(The cancer genome atlas,TCGA)的12种癌症类型,共3 281个样本,进行了突变网络分析,鉴别出16个具有显著突变的子网络,其中包括众所周知的癌症信号传导通路,以及在癌症中具有较少特征作用的子网络;此外,还发现在许多突变子网中,样本突变表现出共发性,即子网的多个基因在多种癌症中均具有体细胞(即非生殖性细胞)突变[29]。
2.3 结合基因组和转录信息的方法
结合基因组和转录信息的方法是利用大量数据库中存放的人类遗传变异数据,多角度全面分析人类疾病(包括癌症)与nsSNVs之间的相关性,揭示疾病的发病机理,进而加快推动有关疾病治疗药物的研发,代表性方法有CADD[30]和Deleterious annotation of genetic variants using neutral networks(DANN)[38]。
Kircher 等人于2014年提出了CADD[30]方法,用以预测nsSNVs致病性。该方法不仅考虑了生物进化保守性特性,而且将转录、调节以及蛋白质水平等信息纳入变异数据特征表示中,使用C-score来衡量编码区和非编码区变异的有害程度;CADD的web server提供了基因组所有86亿个可能SNVs的C-scores和indels评分等[39]。
Quang等人认为CADD[30]使用的支持向量机(Support vector machine,SVM)分类器无法捕获特征间的非线性关系,限制了CADD预测性能的发挥。鉴于此,Quang等人于2015年对CADD进行了改进,使用与CADD相同的样本数据和特征集合,训练深度神经网络,提出了DANN[38],该网络模型比SVM更适用于大样本问题,能够有效捕获特征间的非线性关系。实验结果表明,相比于CADD,DANN的错误率减少了19%,Area under the curve(AUC)增加了14%。
2.4 基于序列进化保守性的方法
基于序列进化保守性的方法,主要通过搜索蛋白质数据库,获得查询序列的同源蛋白质多序列比对结果,根据不同的计算准则,得到该查询序列特定位置处的氨基酸进化保守性,代表性方法有SIFT[31]和Mutation Assessor[40]。
SIFT是通过计算序列中每个氨基酸残基突变为其它类型的可能性,并将序列划分为保守区和非保守区,在保守区的突变倾向于有害突变,在非保守区的突变倾向于中性突变[31]。SIFT不仅可以预测天然的错义突变,还可以预测基于实验室条件下的诱导突变[31]。2018年3月,在SIFT 的web server中,更新添加了SIFT for Genomes和indels预测工具[41]。
Reva等人认为进化保守模式,能够有效地整合氨基酸残基的任何突变影响信息[40],他们开发的Mutation Assessor[40]将序列比对结果进行聚类,然后将聚类结果转化为同源序列中家族和亚家族的保守模式,使用组合熵测量其家族及亚家族中氨基酸残基的进化保守性。他们认为在任何种类保守模式中的残基发生突变,都有可能影响蛋白质功能[40]。
PROVEAN[18]也是一种利用多序列比对预测蛋白质中单个或多个氨基酸突变危害性的方法。与Mutation Assessor[40]与SIFT[31]不同的是,PROVEAN不仅计算感兴趣位置的氨基酸残基,而且衡量邻近侧翼序列的比对质量,使用基于区域的δ比对评分(Delta alignment score,DAS)来预测突变是否对人体产生危害[18]。在2015年1月的更新中,PROVEAN预测了Ensembl v66人类蛋白质中所有可能的SNVs和indels[42]。
已有的非编码区突变预测工具CADD[30]与FATHMM-MKL[43],本地运行均需要大量预先计算的信息[44],为了简化计算,便于预测,Capriotti等人于2017年开发PhD-SNPg[44],仅使用基于序列特征(25个来自于序列编码,10个来自于PhyloP[45]的保守分值),以0到1之间的概率值作为预测输出,大于0.5则认为突变对人体是有害的。
2.5 基于序列和结构混合特征的方法
蛋白质功能和疾病之间的关系错综复杂,当目标蛋白质与环境分子之间有复杂相互作用时,仅依靠统计数据和进化统计分析,预测方法的性能往往受到限制[11]。目前更多的方法是基于序列和结构混合特征进行预测,该类方法通常与机器学习相结合。即利用从序列和结构中获取的特征,训练机器学习分类器模型,预测变异是否对人体产生危害。代表性方法有PolyPhen-2[32]和Functional analysis through hidden Markov models v2.3(FATHMM)[46]。
PolyPhen-2[32]除使用传统特征外,还计算了蛋白质氨基酸残基接触信息,如与杂原子接触(Contacts with heteroatoms),链间接触(Interchain contacts)以及与功能位点接触(如BINDING、ACT_SITE、LIPID以及METAL等),PolyPhen-2将这些功能属性作为突变数据的特征标识,使用Naïve Bayes模型进行预测[32]。在预测错义突变时,其输出结果与几种常见的预测方法不同,SIFT[31],PROVEAN[18]和FATHMM[47]的预测结果为“Neutral”和“Deleterious”两种类别,而PolyPhen-2预测包括“benign”、“possibly damaging”和“probably damaging”等3种结果[32]。
FATHMM[46]利用隐马尔可夫模型(Hidden Markov models,HMM)计算序列的保守性,预测蛋白质错义突变的功能效应。它不仅可以预测编码区的nsSNVs,还可以对非编码变异(Non-coding variants,ncV)进行预测[46]。在FATHMM的nsSNVs致病性预测工具中,包括基于序列保守的加权和未加权两种类别,其中加权算法加入了序列保守性和致病性权重,可以解释序列对变异的耐受性[47]。
CRAVAT[48]、VEST[49]以及SNAP[50]也是基于序列和结构混合特征的nsSNVs致病性预测方法。已有研究表明,nsSNVs的危害性与其周边的微环境息息相关[51,52]。CRAVAT[48]和VEST[49]将变异的序列微环境和局部蛋白质结构性质,作为样本数据的特征标识,即提取氨基酸残基的物理化学性质、蛋白质或DNA多序列比对得分、基于区域的氨基酸序列组成、预测的局部蛋白质结构性质等86个特征,再使用机器学习分类器进行预测[48,49]。SNAP[50]通过计算基于多序列比对的进化保守性得分,结合蛋白质三维结构信息,使用神经网络模型来预测蛋白质序列中的突变,并将预测结果转化为0(Neutral,中性突变)和1(Deleterious,有害突变)两种类别[50]。
2.6 综合评价类方法
综合评价类方法利用多个nsSNVs致病性预测工具的输出结果,设定综合评价得分准则,结合机器学习技术,预测nsSNVs的致病性。综合评价类方法的预测结果通常优于单一预测工具[12,53],代表性方法有Condel[33]和PredictSNP1[12]。
Condel[33]最初整合了5种预测工具(即SIFT[31]、Polyphen-2[32]、MAPP[54]、LogR Pfam E-value[55]和Mutation Assessor[40])的输出结果,通过加权平均计算共有的有害性评分,并将其作为nsSNVs致病性预测的依据。在Condel最近一次更新中,确定Mutation Assessor[40]和 FATHMM[47]组合能够得到的预测结果。在Condel的web server中,提供了预先计算的人类蛋白质全部编码基因的5种工具(Condel[33]、SIFT[31]、PolyPhen-2[32]、Mutation Assessor[40]以及FATHMM[47])的预测结果。
与Condel[33]类似,TransFIC[47]也是一种转换蛋白质突变功能影响评分的预测工具。TransFIC[47]整合SIFT[31]、Polyphen-2[32]和Mutation Assessor[40]的预测得分,将该分数与种系中具有相似功能注释的基因SNVs的分数分布进行比较,并使用Z-score来转换得分,结果表明,对种系SNVs耐受性较差的基因突变得分被增大,相对耐受较好的基因突变得分被降低[47]。TransFIC[47]的预测结果能够提供TransFIC得分、基于SIFT的TFIC_SIFT得分、基于PolyPhen-2的TFIC_PPH2得分以及基于Mutation Assessor的TFIC_MA得分。与整合的3种预测工具相对应,TransFIC还给出3种输出标签:TFIC_SIFT_LABEL、TFIC_PPH2_LABEL和TFIC_MA_LABEL。经过转换后,TransFIC最终用0、1和2代表突变的3种危害程度(Low、Medium和High),分值越大,突变的危害性越大[47]。
PredictSNP1[12]对8种已有的预测工具(MAPP[54]、nsSNPAnalyzer[56]、PANTHER[57]、PhD-SNP[58]、PolyPhen-1[59]、PolyPhen-2[32]、SIFT[31]和SNAP[50])进行无偏估计,并将上述8种预测工具中表现最佳的6种工具,组合成一致性分类器PredictSNP1,其web server提供了上述9种工具的预测结果。
PredictSNP2[60]构建了覆盖不同类别(包括Regulatory、Splicing、Missense、Synonymous和Nonsense variants)的疾病相关变异预测模型,综合了6种工具(CADD[30]、DANN[38]、FATHMM[43]、FitCons[61]、FunSeq2[62]和GWAVA[63])的预测结果,将表现最好的5种工具的输出结果,转化为PredictSNP2共识评分,其web server提供了上述7种工具的预测得分。
REVEL[53]集成了13种预测方法(MutPred[64]、FATHMM v2.3[47]、VEST 3.0[65]、PolyPhen-2[32]、SIFT[19]、PROVEAN[18]、Mutation Assessor[40]、Mutation Taster[66]、LRT[67]、GERP++[68]、SiPhy[69]、phyloP[45]和phastCons[70])的输出结果。REVEL[53]实验结果表明,其优于上述13种单一预测方法,同时也优于7种类似的集成预测方法(MetaSVM[71]、MetaLR[71]、KGGSeq[72]、Condel[33]、CADD v1.3[30]、DANN[38]和Eigen[73])。
3 常见的SNVs数据库
全世界范围内,大量SNVs数据库存储了多态性变异、致病性变异、致癌性突变、与癌症有因果关系的突变基因目录、专家策划的体细胞突变等数据。在nsSNVs致病性预测研究中,这些数据库经常被使用到,下面对6种常见的数据库进行详细介绍。
3.1 intOGen
2018年,Tamborero D等人将已识别的癌症驱动基因,基因测序技术和临床药物反应3者结合,建立了癌症基因组解释器(Cancer genome interpreter,CGI)[74]。CGI不仅可以识别新测序肿瘤的所有已知和可能的致癌基因组突变(包括单点突变、短插入/删除、拷贝数改变和/或基因融合),评估未知意义变体,而且还能注释肿瘤的所有变体,这些变体将构成使用不同临床药物反应的最新生物标志物[74]。
BBGLab使用CGI工具,开发了肿瘤基因组学数据库Integrative onco genomics(intOGen,https://www.intogen.org/search)[75],通过分析66种肿瘤类型,发现并存储了568个癌症驱动基因(Cancer driver gene,CDG)。用户可通过网站查询某个基因在特定癌症中的突变信息(如某种癌症驱动基因或突变频率等),此外,用户还可以下载全部癌症驱动基因数据[75]。
3.2 COSMIC
癌症中的体细胞突变目录数据库(Catalogue of somatic mutations in cancer,COSMIC,https://cancer.sanger.ac.uk/cosmic/)[76],是一个探索体细胞突变对人类癌症影响最大、最全的数据库。它由4个不同子项目组成,每个项目呈现出一个单独的数据集或视图,具体包括:(1)专家策划的体细胞突变数据库COSMIC;(2)超过1 000种用于癌症研究的细胞系突变谱Cell lines project;(3)基于三维结构的癌症突变交互式视图COSMIC-3D[77];(4)具有与癌症有因果关系的突变基因目录Cancer gene census[76]。COSMIC中可以查询基因突变、癌症驱动基因等信息,同时提供数据下载功能,可下载数据包括:所有基因突变数据、基因普查数据及相应突变、拷贝数变异数据、非编码区突变数据以及COSMIC所有基因的fasta格式数据等[76]。
3.3 CanProVar 2.0
以fasta格式存储的人类癌症蛋白质组变异数据库(Human cancer proteome variation database,CanProVar2.0,http://canprovar2.zhang-lab.org/datadownload.php)[3],将变异信息显示在每条记录的头部:“rs”表示变异呈现单核苷酸多态性(Single nucleotide polymorphism,SNP),“cs”表示突变与癌症相关(Cancer related mutations,CAM)。在CanProVar 2.0中,共记录了967 017个SNP和156 671个CAM。此外,该数据库按照癌症类型划分为41子单元,如脑癌(含327个突变)、食道癌(含2 246个突变)、肾上腺肿瘤(含15个突变)等[3]。
3.4 humsavar
人类多态性与疾病突变(Human polymorphisms and disease mutations,humsavar,https://www.uniprot.org/docs/humsavar)数据库[26]中列出了人类UniProtKB/Swiss-Prot[78]中注释的所有错义突变,存储了31 132个与疾病(Disease)相关突变,39 464个多态性(Polymorphism)变异,以及8 383个未标记(Unclassified)变异。值得注意的是,该数据库提供的变异分类仅用于科学研究,而不用于临床和疾病诊断。
3.5 ClinVar
Clinical variants(ClinVar,https://www.ncbi.nlm.nih.gov/clinvar)[25]汇总了来自National Coalition Building Institute(NCBI)的基因组变异,以及这些变异与人类健康间关系的数据库。在ClinVar的搜索栏输入cancer,可以得到数据库中关于癌症的突变分类结果,如针对临床意义,变异分为良性、可能良性、可能致病、不确定意义等7种类别;针对变异对分子产生的后果,可分为移码、错义、拼接、非编码RNA等7种类别;针对变异类型,又包括删除、复制、插入/删除、单核苷酸变异等;除此之外,ClinVar还提供包括变异长度、审核状态、等位基因起源、变异基因关系等角度的突变分类结果数据[25]。
3.6 1 000 Genomes
1000人基因组计划1000 Genomes(http://browser.1000genomes.org)包括了1 000个人(来自于26个人种)的全部基因组数据,以及从数据库的基因组覆盖率和靶向测序中发现的变异信息[79]。用户通过专用的浏览器(1000 genomes browser),可在基因组注释的背景下查看1 000个人基因组数据,如蛋白质编码基因,全基因组调控信息等。1000 genomes browser中还包括所有非同义突变效应预测器(Variant effect predictor,VEP)[80]、SIFT[31]和PolyPhen[32]预测结果。
4 nsSNVs致病性预测常用的特征表示方法
常见的nsSNVs致病性预测方法中,基于序列特征的预测方法有CHASM[81]、PROVEAN[18]、FATHMM[47]、Mutation Assessor[40]和PANTHER[57];基于结构特征的预测方法有SDM[82]和APOGEE[83];目前更多的方法是基于序列和结构混合特征进行预测的,如SNAP[50]、PolyPhen-2[32]和iFish[84];另外,综合评价类方法的特征来自于已有预测方法的输出结果,如Condel[33]、PredictSNP1[12]和REVEL[53]。借鉴上述预测方法所使用的特征,将突变的常用特征表示分为4种类别:(1)基于蛋白质序列的特征;(2)基于蛋白质结构的特征;(3)突变位点微环境特征;(4)基于已有预测工具输出的特征。4类特征表示方法可归纳如下。
4.1 基于蛋白质序列的特征
4.1.1 位置特异性得分矩阵
人类nsSNVs是否对人体产生危害,与生物进化过程息息相关。在蛋白质相关研究中,位置特异性得分矩阵(Position specific scoring matrix,PSSM)包含了非常重要的生物进化信息,已被证明是一种非常有效的特征[85,86]。通常使用Position-specific iterated basic local alignment search tool(PSI-BLAST)[87]工具,搜索Swiss-Port[88]数据库,为输入的待查询蛋白质序列构建同源蛋白质多序列比对(Multiple sequence alignment,MSA),在MSA的基础上计算PSSM信息。
4.1.2 功能位点突变
已有研究表明,突变是否发生在重要功能位点,对突变后蛋白质功能能否正常发挥有较大影响[32,89]。例如,SAPRED[89]通过查询Swiss-Prot[88]数据库,确定突变是否发生在被注释为ACT_SITE以及METAL等功能位点;PolyPhen-2[32]通过查询蛋白质UniProtKB/Swiss-Prot[78]数据库,确定突变是否发生在DISULFID、SIGNAL、BINDING以及ACT_SITE等重要功能位点。在nsSNVs致病性预测研究中,尤其是蛋白质SNVs的研究中,通常会将突变是否发生在活性位点(Active sites)、结合位点(Binding sites)以及非球状区视为特征的重要组成部分。
4.1.3 突变点位性质
(1)物理化学/生物化学性质。在nsSNVs致病性预测研究中,许多文献将蛋白质突变位点的物理化学/生物化学性质作为特征的重要组成部分,如突变前后氨基酸亲水性(Hydrophilicity)、疏水性(Hydrophobicity)、体积(Volume)、分子量(Molecular weight)、电极性(Polarity)等属性值以及突变前后变化值[2,11,90]。
氨基酸的物理化学/生物化学属性值可通过氨基酸索引数据库(Amino acid index database,AAindex,https://www.genome.jp/aaindex/)[91]查询得到。AAindex[91]是一个数字索引数据库,代表氨基酸和氨基酸对的各种物理化学和生物化学特性,所有数据均来自于已发表文献;它由AAindex1(代表20个数值的氨基酸索引)、AAindex2(代表氨基酸突变矩阵)和AAindex3(代表统计蛋白质接触电位)等3个部分组成。在最近更新的版本(v9.2,Feb 2017 Released)中,AAindex1共存储了566种氨基酸指标。
(2)替换打分矩阵BLOSUM和PAM。BLOSUM[92]和PAM[93]是蛋白质序列比对的替换打分矩阵,用于计算任意两条序列的相似性,发现两者的生物进化关系,进而有效地分析和预测基因功能。例如,使用PSI-BLAST[87]计算PSSM信息时,默认的替换打分矩阵是BLOSUM62[92]。在nsSNVs致病性预测研究中,通过查询此两类矩阵,可以得到突变氨基酸对的替换打分值,并将其归为突变样本的特征组成部分。
4.2 基于蛋白质结构的特征
基于蛋白质结构的特征分为两类:(1)蛋白质在Protein data bank(PDB)[94]中有已知的三级结构,可直接提取结构信息;(2)蛋白质三级结构未知,但可通过计算机软件模拟的方式得到蛋白质结构信息。
4.2.1 蛋白质三级结构已知
若蛋白质在PDB[94]中存在已知的三级结构,则可利用Dictionary of secondary structure in proteins(DSSP)[95]、Biopython中的The PDB Module[96]等工具,获得关于突变位点的二级结构(Secondary structure,SS)、溶剂可及表面积(Solvent accessible surface area,SASA)、无序区域(Disorder region,DR)、Phi-Psi二面角、原子的空间坐标(Atomic spatial coordinates)等结构信息[94],并将这些信息作为突变样本的特征组成部分。
4.2.2 蛋白质三级结构未知
对于三级结构未知的蛋白质,可通过计算软件预测的方式获取结构信息,并将这些预测信息纳入突变样本的特征表示中,丰富特征的同时,以期提高nsSNVs致病性预测的准确度。以下是几类常见的结构信息预测方法。
(1)蛋白质三维结构预测。由Zhang Lab研发的Iterative threading assembly refinement(I-TASSER)[97]和QUARK[98]是全球领先的蛋白质三级结构预测工具。截至2020年9月25日,已有来自149个国家的136 217个使用者,利用I-TASSER的web server预测了571 261个蛋白质的结构。I-TASSER[97]的返回结果包括预测的二级结构及对应得分、预测的溶剂可及性面积、预测的标准化B因子、I-TASSER使用的前10个threading模板以及预测的Top 5模型等信息[99]。在nsSNVs致病性预测研究中,可以提取I-TASSER预测的部分相关结构信息,将其作为突变样本的特征组成部分。
(2)相对溶剂可及性预测。已经有多种方法可以用于预测蛋白质的相对溶剂性(Relative solvent accessibility)。SANN[100]是此类方法的一个代表,它为蛋白质序列中每个氨基酸残基提供3种概率值,即分别属于埋藏(Buried)、中间(Intermediate)和暴露(Exposed)的概率值[100]。
(3)二级结构预测。预测得到的二级结构(Predicted secondary structure,PSS)亦是一类非常有效的结构特征[101,102],可以通过PSIPRED[103]工具预测获得。该工具为蛋白质序列中的每个氨基酸残基提供3个概率值,即分别属于Coil(C)、Helix(H)和Strand(S)的概率值[103]。
(4)无序区域预测。蛋白质的无序区域(Disorder)是指不具有固定的三级结构,部分或完全展开的区域。该区域被认为参与许多重要功能,如DNA识别、特异性调节等[104]。已有研究表明,在无序区域中发生的突变,会对蛋白质的功能产生影响[105]。蛋白质的无序区域可通过DISOPRED[106]等多种软件预测得到。
4.3 突变位点微环境特征
突变位点的微环境,可以由基于蛋白质三维结构的“邻居”(与突变位点空间距离小于特定Å范围)氨基酸残基构成,也可以由基于蛋白质序列的“邻居”(与突变位点在序列上小于特定长度范围)氨基酸残基构成[94]。文献研究结果表明,单个氨基酸残基突变是否有害,通常与其微环境中氨基酸残基相关[51,52,89]。因此,在包括nsSNVs致病性预测在内的蛋白质相关研究中,通常会提取微环境范围内信息,并将其作为待研究问题的特征组成部分,以期丰富特征表示,提高预测性能[107]。在提取微环境特征时,滑动窗口大小的确定是至关重要的一步[108]。
4.4 基于已有预测工具输出的特征
综合评价类工具Condel[33]、TransFIC[47]、PredictSNP1[12]、PredictSNP2[60]和REVEL[70]的web server提供了多种工具的突变预测结果。例如,Condel[33]提供了预先计算的所有人类蛋白质编码基因中全部可能变异的Condel[33]、SIFT[31]、PolyPhen-2[32]、Mutation Assessor[40]和FATHMM[46]5种预测得分[47];TransFIC[47]提供突变数据的TransFIC、TFIC_SIFT、TFIC_PPH2以及TFIC_MA 4种预测得分[47];PredictSNP1[12]提供突变数据的MAPP[54]、nsSNPAnalyzer[56]、PANTHER[57]、SIFT[31]以及SNAP[50]等9种预测得分。借鉴综合评价类方法的思想,将已有工具的预测得分,作为突变样本的特征表示,连同提取的其它特征,设定综合评价得分准则,结合机器学习技术,对nsSNVs的致病性进行预测。
5 突变预测结果评价
混淆矩阵(Confusion matrix)由真阳性(True positive,TP)、真阴性(True negative,TN)、假阳性(False positive,FP)和假阴性(False negative,FN)组成。基于上述4个值,衍生出一系列评价指标,如准确度(Accuracy,ACC)[109]、马修斯相关系数(Matthews correlation coefficient,MCC)等[110,111]。ACC是预测正确的样本数(包括真阳性和真阴性)占所有样本数的比例。当正负样本数量严重不均衡时,MCC被认为是衡量混淆矩阵的最佳指标,其取值范围是-1到1,其中1表示预测值与真值完全相同,0表示预测结果随机,-1表示完全相反。通常,MCC的值越大,分类效果越好。具体公式如下
(1)

(2)
除ACC和MCC之外,Receiver operating characteristic(ROC)和AUC也是重要的分类效果评价指标。ROC曲线的x轴是假阳性率(False positive rate,FPR),表示预测为假阳性样本数占所有真阴性样本数的比例,y轴是真阳性率(True positive rate,TPR),表示预测为真阳性样本数占所有阳性样本的比例,这是两个相互独立的统计量。对于一个预测结果为概率值的分类器,可以设置若干个阈值θ,每个θ对应一组(FPR,TPR),由此绘制ROC曲线[112]。ROC曲线越接近左上角,表示分类效果越好。AUC是ROC曲线下的面积,通常认为面积越大,分类效果越好[113]。
6 12种常见nsSNVs致病性预测方法比较
机器学习技术在蛋白质相关研究领域有着广泛的应用,如蛋白质配体绑定预测[107,114]、跨膜蛋白预测等[115,116]、DNA配体绑定位点预测[117]、蛋白质三级结构预测[118]、蛋白质残基接触图预测[119]、nsSNVs致病性预测[32,43]等。表2列出了12种常见的nsSNVs致病性预测方法的比较结果。分析表2中的数据,可以得到如下结论。
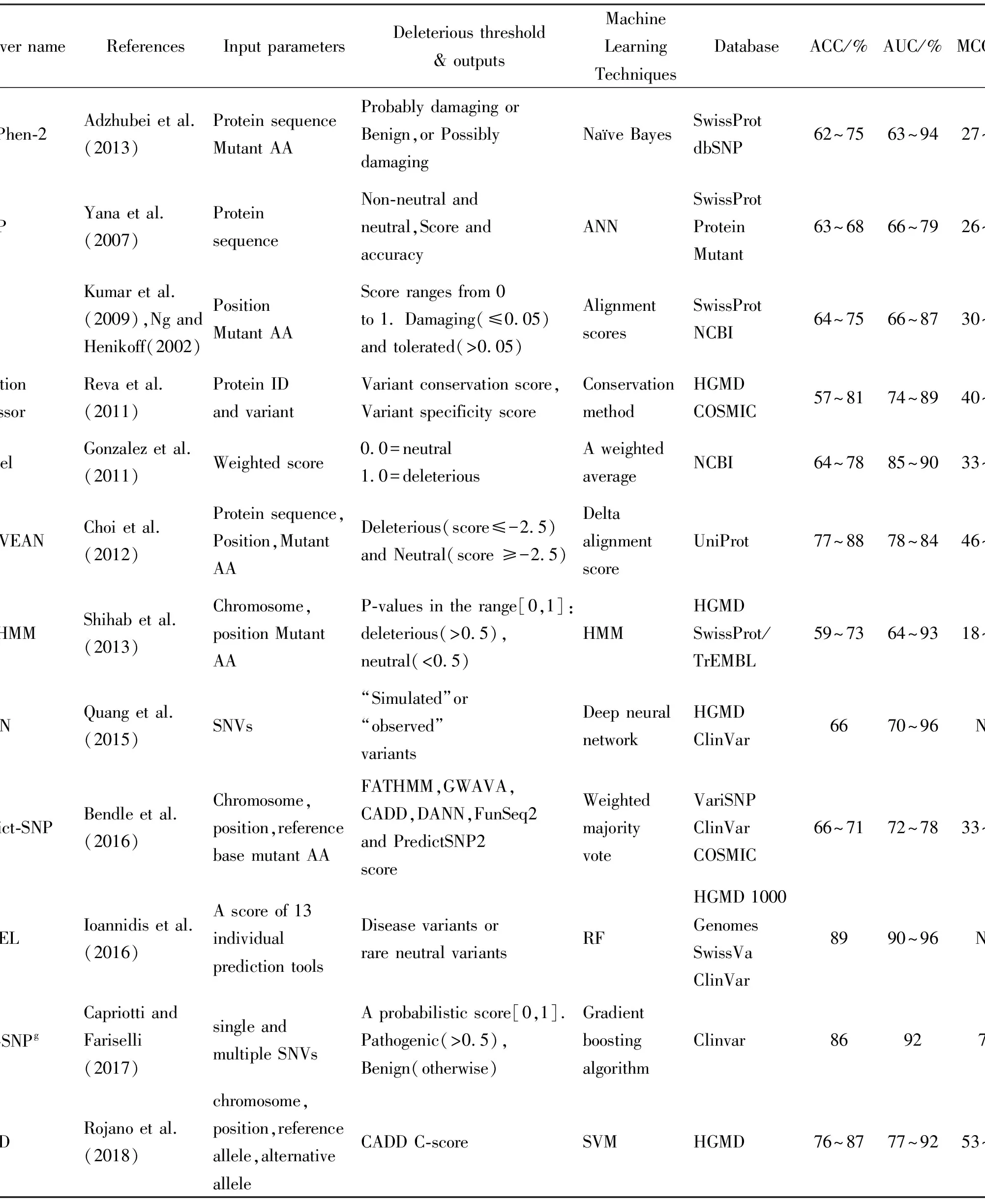
表2 12种常见nsSNVs致病性预测方法的比较(数据来自于文献[1])
(1)经典预测工具SIFT[31]、PROVEAN[18]、PolyPhen-2[32]和CADD[18]具有较高的MCC。其中,SIFT[31]和PROVEAN[18]是基于序列进化保守性的方法,构建查询序列的同源蛋白质多序列对比是其核心思想。PolyPhen-2[32]将提取的序列和结构混合特征作为输入,训练Naïve Bayes模型,最终的错义突变预测结果包括“benign”、“possibly damaging”和“probably damaging”等3种类别,这与只有2类别预测结果的方法(SIFT[31]、PROVEAN[18]、Mutation Assessor[40]等)有较大不同。CADD[18]为每个变异提取了949个特征,使用SVM分类器来捕获变异特征间的线性关系,对变异有害性进行预测。
(2)REVEL[53]和DANN[38]具有较高的AUC。REVEL[53]是一种综合评价类工具,它集成了13种单一预测方法的输出结果,其预测性能优于13种单一预测工具,同时也优于其它7种类似的综合评价类预测方法[53]。DANN[38]使用了与CADD[18]相同的数据集和特征,研究结果表明,基于深度学习框架的DANN[38]能更有效地捕获特征间的非线性关系。
(3)总结相关文献,发现随机森林决策树(Random forest,RF),SVM以及基于Voting的机器学习方法,在不同基准数据集的变异预测方面均表现出较优秀的预测效果[1]。
7 结束语
随着nsSNVs对人类健康影响研究的快速开展,截至2020年9月,intOgen记录了66种癌症的203 003 820个突变信息[75];COSMIC的癌症基因普查数据库中记录的致癌基因共723个[76];CanProVar 2.0中记录了156 671个蛋白质SNVs与癌症相关[3];humsavar中记录了39 464个呈现多态性的nsSNVs[26]。
该文对nsSNVs致病性预测的5个方面进行了概述,包括nsSNVs变异类别、致病性预测方法、常见SNVs数据库、特征表示和常见nsSNVs致病性预测方法比较。在致病性预测方法分类方面,根据方法所使用特征的不同,将本领域的国内外研究归纳为6大类别:基于突变频率的方法需要大量的先验知识,主要根据突变频率在中性和致病性样本间的差异进行预测,但此类方法难以鉴别具有低复发频率的驱动基因;基于通路的方法考虑突变基因间的相互作用关系网络以及生物效应,是一类非常有效的驱动突变预测方法;结合基因组和转录信息的方法充分利用了人类基因组测序得到的数据,能够更全面地解释疾病发病机理;基于序列进化保守性,以及基于序列和结构混合特征的预测方法是较为常见的,代表性方法有SIFT[31]和PolyPhen-2[32]等;综合评价类方法是根据若干已有预测工具的输出,制定打分准则,综合评价,此类方法的效果往往优于单一预测工具。
尽管目前进行了很多nsSNVs致病性预测研究,但快速准确地预测突变是否会导致疾病(或癌症),仍是一项非常具有挑战性的工作,以下几个方面可能是潜在的研究突破点。
(1)基于全基因组数据的预测。从DNA微阵列技术测得信使核糖核酸(Messenger ribonucleic acid,mRNA)的表达丰度,到转录调控通路,蛋白质翻译,再到染色体结构,从这一系列过程中,发现突变的根源,挖掘基因突变在后续转录、调控、翻译等过程的系列影响,揭示突变对蛋白质功能效应的影响机制,加快寻找致癌性突变的靶向药物。
(2)深度学习框架的引入。人类基因组数据庞大,需要综合考虑的因素错综复杂,数据样本通常以万为单位计量,深度学习框架或许能够解决数据量大,同时满足预测准确率高的要求。
(3)更多临床实时数据的加入。癌症驱动基因的预测和发现,对指导癌症临床的早期诊断和药物靶向治疗有着重大意义;与此同时,临床癌症样本、药物反应等实时数据,对理论研究也有较大的推动作用。
猜你喜欢
今日农业(2021年11期)2021-08-13 08:53:24
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
湖南畜牧兽医(2016年3期)2016-06-05 08:37:56
兽医导刊(2016年12期)2016-05-17 03:51:42
百科知识(2015年18期)2015-09-10 07:22:44
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
当代畜禽养殖业(2014年7期)2014-02-27 07:59:20
现代检验医学杂志(2014年1期)2014-02-06 01:29:25
