
高效率蓝光“热”激子材料的分子设计*
2021-03-05 10:04潘玉钰
广州化工 2021年4期
赵 梅,潘玉钰
(沈阳工业大学石油化工学院,辽宁 辽阳 111003)
有机电致发光器件(OLED)经过近三十年的发展,在材料制备、工艺优化及相关理论研究方面都取得了巨大进步;其中, 有机发光材料作为OLED的核心技术,已成为该领域国际竞争的焦点[1-6]. 有机电致发光过程是:注入的空穴和电子复合形成激子,激子经辐射跃迁到基态(S0)而发光。一般三重态(T)激子由于自旋禁阻几乎无法辐射跃迁到单重基态(S),导致占总数75%的三重态激子几乎完全浪费。故传统的荧光材料由于仅仅单重态激子发光,存在内量子效率为25%的理论上限,能量利用效率低[7-8]。磷光材料中,由于重金属原子增强自旋-轨道耦合,促进单重态和三重态的系间窜越,使三重态激子可以直接辐射跃迁回到S0态发光,内量子效率理论上限可达100%,即单重态和三重态激子同时发光[9-11]。但磷光材料存在蓝光材料稀少,重金属价格昂贵等缺点。那么,如何在没有金属原子的全有机荧光材料体系中实现100%的激子利用?唯一可行的方法是把75%非辐射的三重态激子通过特殊途径转化为可辐射的单重态激子。
2013年,华南理工大学马於光教授研究组设计的“热”激子(“Hot” exciton)通道材料,理论上可以同时实现最大化激子利用和最大化激子辐射,是一类具有自主知识产权,有广阔应用前景的新材料[12-15]。
事实上,理想的“热”激子型分子结构和激发态分布存在三条基本原则:(1)分子至少具有可被调节的两部分,即给体-受体(D-A)结构,而且D-A 间相互作用不宜太强-避免电荷转移态(CT态)成为最低激发态造成较低的荧光效率,也不宜太弱-CT态不能有效形成。(2)发光态(S1)应以局域态(LE)为主,保证 S1态高的荧光效率;另一方面则是保持高能级的Sn(n>1)和Tm(m>1)具有显著 CT 激发态特征,实现足够小的单重态与三重态的能量差(ΔESn-Tm),快速的完成激子反向系间穿越(reverse intersystem crossing,RISC)RISC (Tm→Sn)过程。(3)负责激子类型转换的高能级Tm激子与Tm-1激子之间具有足够大的能隙(ΔETm-Tm-1),从而有效的降低内转换过程IC (Tm→Tm-1)速率,使RISC的速率足以与之抗衡竞争,最终导致T 激子的弛豫路径发生改变,偏离T1态;这样也能够有效避免了T1激子的生成和积累,克服有机磷光材料和TADF材料的T-T湮灭导致器件在高电流密度下效率滚降严重的问题。
在前期的工作中我们报道了蓝光分子TPA-AN (4-(anthracen-9-yl)-N,N-diphenylaniline ), 此分子为典型的D-A结构,电子给体部分为三苯胺(TPA),电子受体部分为蒽 (AN),D与A部分以单键连接。经过实验测定TPA-AN分子的薄膜发光效率可以达到50%,掺杂器件的内量子效率可以达到30%左右[16]。由此可见,此分子虽然突破内量子效率25%的理论上限但较低的还不足以达到商用的标准。分析原因得出,作为“热”激子材料,此分子兼具所有上文提到的条件,但分子激发态中具有CT态的能级处于S3态和T8态,特别是三重态所处能级较高,这可能是导致激子不能有效的RISC的主要原因[17]。
本文,我们基于理想的“热”激子型材料的设计原则,对TPA-AN分子进行结构优化,以AN为发光核分别引入电子性质不同的基团,通过理论模拟对其构型和激发态性质进行分析,对其光电性质实现改性,达到兼具高量子效率和高辐射效率的“热”激子型分子。
1 计算方法的选择
目前,分子模拟的方法众多,但在发光材料激发态模拟计算方面TD-DFT方法是应用最广、性价比做高的方法[18-20]。但众所周知,TD-DFT方法在激发态的计算过程中泛函依赖性较大,尤其对于分子中具有CT态的激发态能量存在低估。因此在计算之前需要进行泛函的选择。在前期的工作中,我们已经对CT态分子进行了计算泛函的选择,经过与实验测得的光谱进行比较发现长程校正泛函ωb97x具有很好的重复性,因此我们在接下来的计算中选用TD-DFT/ωb97x/6-31G (d,p)方法进行计算[21]。计算所用软件为Gaussian 09.D. 01[22]。
2 结果与讨论
2.1 分子设计
为保留分子的蓝光性质以及三重态中较大的能级差,我们选取了经典蓝光材料AN作为发光核,为避免分子间作用力过大我们在AN分子两侧分别加入苯环后与X部分和D部分以单键相连[23-25]。分子结构如图2所示。
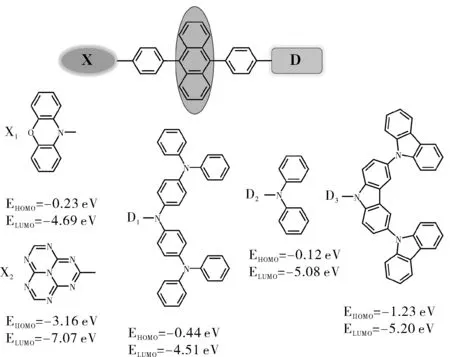
图1 分子结构示意图Fig.1 Molecular structure
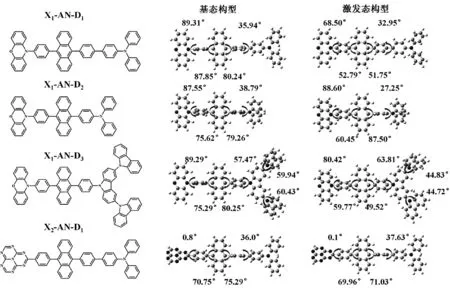

图2 基态与激发态下分子的优化构型Fig.2 Optimal configuration of molecules in ground state and excited state
其中X部分我们分别选取电子给体基团吩噁嗪作为X1,电子受体基团1,3,3a1,4,6,7,9-七氮杂菲作为X2,D部分我们依据基团的最高占据轨道能量 (EHOMO) 不同,筛选了具有不同给电子能力D1(三苯胺),D2(二苯胺),D3(9,3′:6′,9″-三咔唑),由此构建出了6个不同的分子:X1-AN-D1, X1-AN-D2, X1-AN-D3和 X2-AN-D1, X2-AN-D2, X2-AN-D3。
2.2 分子基态和激发态构型
首先,我们将这六个分子分别在真空中进行了基态和激发态的构型优化。从基态构型我们可以看出,AN与之相连的苯环之间都存在较大的扭曲角,这种较大的扭曲角有利于发光态的局域发光,增强分子的发光效率。而苯环连接不同的X基团或D基团时扭曲角的变化较大。例如,苯环与吩噁嗪连接时其扭曲角都在89°左右;与1,3,3a1,4,6,7,9-七氮杂菲相连时,由于位阻几乎为零,因此它们之间的二面角几乎为0°;当苯环与三苯胺中的另一个苯环或者与二苯胺中的N相连时,其间的扭曲角集中在35°~38°之间,而与咔唑相连时二面角则增加到了57°左右。这些不同范围的扭曲角为分子激发态的构型弛豫提供了很大的自由度。我们随后对这6个分子进行了激发态的构型优化,从图3中可以看出,与X基团和D基团相连的扭曲角在激发态状态下并没有发生明显的弛豫,二面角变化不大;而AN与苯环之间的扭曲角在激发态下弛豫现象明显,二面角有了10°~20°的减小,这为激发态电子云的离域提供了可能。
2.3 分子的激发态性质
2.3.1 吸收发射光谱分析
首先对分子的吸收(UV)和发射(PL)光谱进行了模拟。
将分子的最大吸收波长(λ/nm)和振子强度(f)列于表1中,为方便比较,将分子TPA-AN的吸收光谱和发射光谱值同时列出。从表1中我们可以看出,虽然在AN周围进行了修饰,但所设计的分子的发光依然在蓝光范围内,发光甚至发生蓝移,并且在发射光谱中普遍表现出较大的振子强度,这说明其可能具有较高的荧光效率。因此从发光性质上看,我们所设计的分子满足较高荧光效率的蓝光发光的要求。
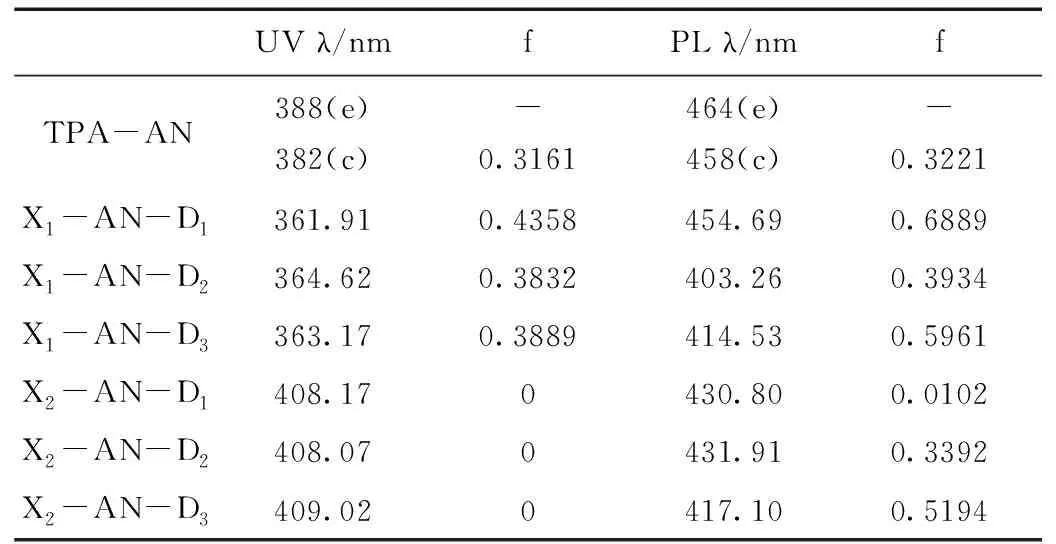
表1 TPA-AN分子和设计分子的吸收与发射光谱最大特征波长(λ)、振子强度(f)数据Table 1 The maximum wavelength (λ) and oscillator intensity (f) data of the absorption and emission spectra of the TPA-AN and designed molecules
2.3.2 激发态能量分析
激发态的跃迁能量变化反映出激发态中尤其是较高能级的激发态的跃迁特征。上文中提到,理想的“热”激子材料须具有较大的ΔETm-Tm-1,从图3可以看出,X1-AN-D1, X1-AN-D2, X1-AN-D3分子的激发态能级中具有较大的ΔET2-T1,但是当X部分换成1,3,3a1,4,6,7,9-七氮杂菲后, T1态能量有了很大的提高,并且T2、T3态能量也有所下降,这导致X2-AN-D1, X2-AN-D2, X2-AN-D3分子中的ΔETm-Tm-1缩小。激发态能量分析说明X1-AN-D1, X1-AN-D2, X1-AN-D3分子有可能构筑出理想的“热”激子材料,因此下面部分我们重点对于这三个分子的性质进行分析。

图3 分子激发态中前10个激发能量图Fig.3 Energy diagram of the first ten singlet and triplet excited-states in molecules
2.3.3 分子跃迁性质分析
对激发态的跃迁性质进行分析。如图4所示我们分别列出了X1-AN-D1, X1-AN-D2, X1-AN-D3分子的前5个激发态的自然跃迁轨道(The natural transition orbital,NTOs)图。NTOs可以体现出跃迁前后的电子云变化。前文中提到造成TPA-AN分子激子利用率较低的原因可能为三重激发态中CT态处于较高能级(T8),导致激子RISC速率无法与激子的IC速率竞争。但如图4所示,三个分子的单重态中S2和S3态为明显的CT态跃迁,值得注意的是,由于分子中引入了电子给体吩噁嗪,三重态跃迁中的CT态能量发生了明显的降低,例如,分子X1-AN-D1中的T3态,X1-AN-D2中的T4态以及X1-AN-D3中的T4和T5态。这种激发态能级的分布特点有利于三重态激子从T3或者T4态通过RISC回到单重态,进而提高激子利用率。
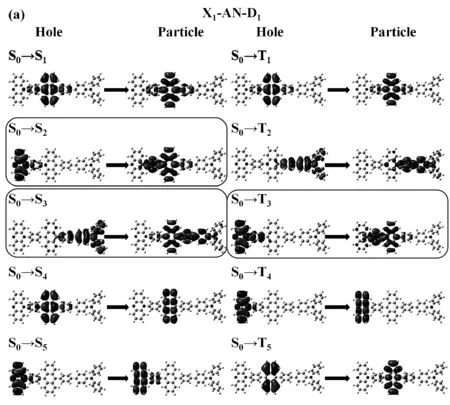
图4 分子中前5个激发态的自然跃迁轨道图Fig.4 The natural transition orbital diagram of the first 5 excited states in the molecules
3 结 论
通过对于蓝光分子AN进行修饰,基于X-AN-D结构,我们设计了6个不同的分子,通过基态、激发态构型分析以及发光性质的探究得出以下结论:
(1)对分子两侧分别引入不同的电子给体和电子受体并没有对光色产生大的影响,一系列分子的发光依然处在蓝光区并普遍具有较高的荧光效率;
(2)激发态能量分析得出,吩噁嗪基团的取代并没有影响构筑“热”激子材料必须的大的ΔETm-Tm-1,为激子反向系间穿越提供了保障;
(3)激发态跃迁性质分析中,由于电子给体吩噁嗪的引入,降低了三重态中CT跃迁态的能量,由分子TPA-AN中的T8态降低到了T3或T4态,这有利于高能级激子通道的构筑;
综上所述,我们所设计的分子X1-AN-D1, X1-AN-D2, X1-AN-D3满足理想型“热”激子材料的全部特征,可能发展成为一类高效率的蓝光“热”激子材料。
猜你喜欢
汕头大学学报(自然科学版)(2020年4期)2020-12-14
物理学报(2019年10期)2019-06-04
铜仁学院学报(2018年6期)2018-07-05
科学之谜(2018年3期)2018-04-09
衡阳师范学院学报(2016年3期)2016-07-10
原子与分子物理学报(2015年3期)2015-11-24
原子与分子物理学报(2015年1期)2015-11-24
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
读写算·教研版(2014年12期)2014-09-01
原子与分子物理学报(2014年1期)2014-03-20
