
新型吲唑衍生物1-乙酸-吲唑-3-羧酸的合成*
2021-03-05 10:04张金龙陶紫薇李直坤刘文俊罗世鹏
广州化工 2021年4期
张金龙,陶紫薇,李直坤,刘文俊,戎 强,罗世鹏
(江苏理工学院化学与环境工程学院,江苏 常州 213001)
作为一种吲哚化合物的等电子体,吲唑化合物具有与吲哚化合物相似的生物活性,如具有避孕、抗炎等药效,还可以作为开发新型抗肿癌药物的基础原料[1-6]。吲唑类化合物是一个重要的医药中间体,目前已有多种吲唑类衍生物已经进入临床研究或上市[7],例如氯尼达明[8]、盐酸格拉司琼[9]等。此外,吲唑羧酸化合物由于同时含有易配位的N原子以及O原子,在与过渡金属或稀土金属配位时,能够得到结构和功能多样的新型配合物,如金属有机骨架材料[10]。通过调控合成,在吲唑氮原子上引入不同的基团,可以多样化地合成各类新颖的配体,以达到合成不同功能金属配合物的目的。本文设计并合成了一个新型的吲唑二羧酸化合物,可以进一步进行生物活性测试,并根据测试结果进行修饰并优化其结构。此外,它兼具吲唑的骨架以及羧酸的配位功能[11],可以作为金属有机骨架配合物的配体,与各类金属盐进行溶剂热等反应,从而制备新颖多样的金属配合物。

图1 两种含吲唑结构的药物分子Fig.1 The chemical structures of two indazole compounds
1 实 验
1.1 主要仪器与试剂
400 MHz核磁共振仪, 德国Bruker;Agilent 6460 ESI-MS质谱仪,美国安捷伦;DF-101S集热时恒温加热磁力搅拌器,河南予华;RE-52A旋转蒸发仪,上海亚荣;AL204分析天平,瑞士梅特勒-托利多。
1H-吲唑-3-羧酸(98%),氯乙酸乙酯(98%),无水碳酸钾(99%),氢氧化钠(97%),安耐吉化学;浓硫酸(GR),无水乙醇(AR),乙腈(AR),乙酸乙酯(AR),国药集团化学试剂有限公司;其它试剂或原料均为市售或化学纯,实验用水为去离子水。
1.2 合成步骤
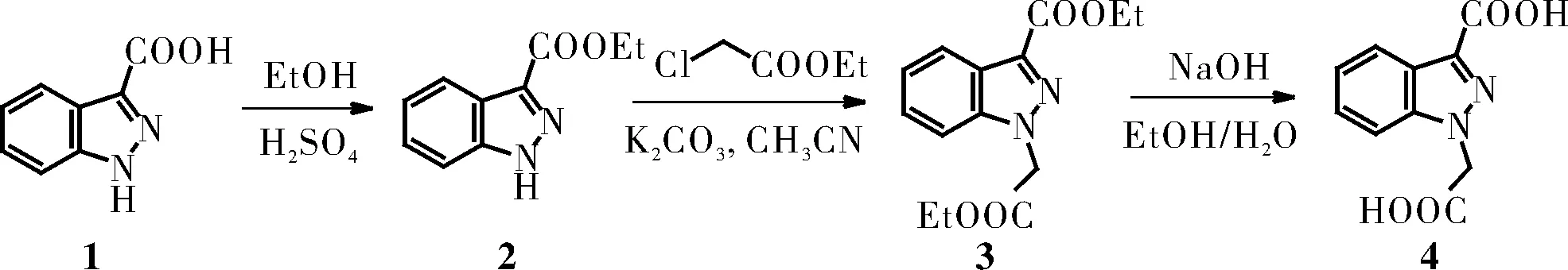
图2 1-乙酸-吲唑-3-羧酸的合成路线Fig.2 Synthetic route of 1-(carboxymethyl)-indoxazole- 3-carboxylic acid
1.2.1 化合物2的合成
称取固体1H-吲唑-3-羧酸(8.1 g,50 mmol)加入带有磁子的250 mL圆底烧瓶之中,然后加入100 mL无水乙醇,在室温搅拌下缓慢滴加5.0 mL浓硫酸,加完后接冷凝管,并置于油浴中回流搅拌4 h。反应结束后减压浓缩除去乙醇,然后在搅拌下加入饱和碳酸氢钠溶液中和反应至pH=7~9。该水溶液适量乙酸乙酯萃取3次。有机相用饱和食盐水洗涤1次,无水硫酸钠干燥,旋干后用乙酸乙酯重结晶,得到黄色固体8.80 g,产率为92.7%。熔点:138~139 ℃;1H NMR (CDCl3, 400 MHz):δ13.19 (s, 1H), 8.28~8.22 (m, 1H), 7.93~7.87 (m, 1H), 7.54~7.47 (m, 1H), 7.41~7.33 (m, 1H), 4.60 (q,J=7.2 Hz, 2H), 1.50 (t,J=7.2 Hz, 3H).13C NMR (CDCl3, 100 MHz):δ163.3, 141.5, 136.3, 127.2, 123.2, 122.3, 121.6, 111.6, 61.1, 14.4. ESI-MS (M+H): 191.2。
1.2.2 化合物3的合成
称取1.92 g (10 mmol) 化合物2以及2.76 g无水碳酸钾(20 mmol)于150 mL烧瓶中,然后再加入25 mL乙腈。于室温搅拌下加入1.61 mL(15 mmol)氯乙酸乙酯,接冷凝管后置于油浴中搅拌回流。薄层色谱跟踪反应,约6 h后原料反应完全,将反应液降至室温,抽滤并用二氯甲烷洗涤滤渣,将母液旋干后用乙酸乙酯重结晶,抽滤,正己烷洗涤后得到白色固体2.42 g,产率87.5%。熔点:76~78 ℃。1H NMR (CDCl3, 400 MHz):δ8.32~8.26 (m, 1H), 8.28~8.22 (m, 1H), 7.56~7.47 (m, 1H), 7.45~7.35 (m, 1H), 5.29 (s, 2H), 4.55 (q,J=7.2 Hz, 2H), 4.24 (q,J=7.2 Hz, 2H), 1.51 (t,J=7.2 Hz, 3H), 1.27 (t,J=7.2 Hz, 3H).13C NMR (CDCl3, 100 MHz):δ167.0, 162.4, 141.2, 136.1, 127.3, 123.6, 123.2, 122.3, 109.3, 61.9, 61.0, 50.9, 14.4, 14.0. ESI-MS (M+H): 277.1。
1.2.3 化合物4的合成
称取5.52 g(20 mmol)化合物3于150 mL烧瓶中,加入40 mL无水乙醇,搅拌溶解,然后加入40 mL 5 mol/L NaOH水溶液,并置于60 ℃ 油浴下反应18 h。待反应结束旋出乙醇,剩余水溶液用盐酸调节pH至2~3,产生大量白色絮状固体,中和完毕后置于4 ℃静置6 h进一步冷却析出白色固体。抽滤,依次用冰水、乙醇洗涤,得到白色固体4.11 g,产率为93.3%,熔点:284~286 ℃。1H NMR (DMSO-d6, 400 MHz):δ8.12~8.08 (m, 1H), 7.63~7.57 (m, 1H), 7.45~7.37 (m, 1H), 7.28~7.22 (m, 1H), 5.03 (s, 2H).13C NMR (DMSO-d6, 100 MHz):δ169.7, 164.7, 141.4, 136.3, 126.4, 123.6, 122.5, 122.0, 111.4, 53.2. ESI-MS (M-H): 219.2。
2 结果与讨论
通过控制变量法,如其它条件不变,改变反应底物的摩尔数之比,反应温度等条件,考察每一步反应的最佳反应条件。结果表明,在化合物2的合成过程中乙醇的含水量对产率有较大影响,反应溶剂最好是经过干燥的无水乙醇,水分较多时会导致产率降低;此外,在中和硫酸的过程中,由于要加入大量的碳酸氢钠,会产生大量气泡,因此要控制加入速度,或采用大口锥形瓶进行中和反应,防止反应溶液随气泡喷溅造成危险和损失。在化合物3的合成过程中,反应温度以及反应摩尔数有较大影响,当氯乙酸乙酯与原料(化合物2)的摩尔数之比超过2.0时,对后期重结晶有影响,不利于产物纯化;而摩尔数比过小时(小于1.2)原料反应不完,综合发现,摩尔数为1.5时可以达到最佳效果;而且反应温度以及反应浓度对反应产率均有较大影响,该步骤的反应浓度为0.5 mol/L时达到最佳,当浓度较小时,原料反应不完,浓度太大时原料无法完全溶解,而反应在回流条件下,能够以较快的速度达到反应完全。在同时水解两个酯基得到二羧酸化合物4的反应步骤中,反应温度应严格控制在60 ℃以达到最佳效果,当反应温度过低时,反应较慢水解不完全,而当反应温度过高(回流)时,吲唑3位的羧基会发生脱羧副反应,无法得到目标产物。
3 结 论
本文以1-氢吲唑-3-羧酸为起始原料,通过在硫酸催化下与无水乙醇化生酯化,然后与氯甲酸乙酯发生取代反应,最后同时将两个酯基水解,以三步76%的总收率得到一个全新的吲唑二羧酸衍生物。本路线简洁高效,纯化步骤简便易行,为后期生物活性测试及作为配体进行金属配合物的制备奠定基础。
猜你喜欢
发明与创新(2022年31期)2022-11-03
中国医药科学(2022年5期)2022-05-05
建材发展导向(2021年24期)2021-02-12
西南军医(2016年6期)2016-01-23
中国继续医学教育(2015年6期)2016-01-07
化工进展(2015年6期)2015-11-13
中国塑料(2015年2期)2015-10-14
中国塑料(2014年1期)2014-10-17
无机化学学报(2014年8期)2014-02-28
无机化学学报(2014年7期)2014-02-28
