
以Eu3+为探针研究吸附状态蛋白质的金属离子结合性质
2021-03-03 06:47荣志江孙红艳郝春燕张跃忠杨斌盛
山西大学学报(自然科学版) 2021年6期
荣志江,孙红艳,郝春燕,张跃忠,杨斌盛
(1.太原科技大学 化学与生物工程学院,山西 太原,030024;2.山西大学 分子科学研究所,山西 太原,030006)
0 引言
蛋白质在不同介质上的吸附行为研究越来越受到广泛关注,其对预防和减少材料表面的蛋白质吸附污染和保证医学移植材料的生理功能等方面具有重要意义[1-2]。由于蛋白质属于两性分子,结构复杂,因此影响蛋白质分子在电极表面吸附的因素很多。除了外界因素,库仑力,憎水作用等还与蛋白质本身的性质有关,如分子量、蛋白质内部稳定性、蛋白质表面荷电性质、在电极表面的三维结构和取向等。一个统一认识是,蛋白质在吸附时构象变化引起的熵增足以弥补静电斥力对吸附的影响[3-6]。蛋白质不同于聚合物,在其复杂性结构的背后,蕴藏着许多生理活性,而关于蛋白质活性和吸附性之间的关系报道的却较少。溶液中游离态中心蛋白结构和功能已被广泛研究,对吸附态中心蛋白基本涉及很少,而蛋白质生物功能的发挥却常常是在吸附状态下完成的。
随着稀土在工业、农业、医药卫生各个领域的广泛应用,稀土元素对环境和人类健康的影响越来越受到重视。课题组使用光学方法已经证明中心蛋白可以与稀土离子相互作用,稀土离子在中心蛋白N端有两个低亲和位点[7]。稀土离子与两个位点结合会引起蛋白质构象变化进而发生聚集,但光谱滴定曲线只出现一个拐点,未能直接区分两个不同的结合位点。应用电化学滴定方法则发现中心蛋白的构象改变能够被滴定曲线敏感体现出来[8]。
来自革兰氏阴性菌质膜氧化空间的CopC蛋白,含有102个氨基酸残基,不含α螺旋,是由9股β折叠片形成的希腊桶状结构[9-10],结构较为致密且蛋白质表面疏水性较强,圆二色研究发现在SDS的作用下有向疏松结构-螺旋化的趋势。CopC可以特异性地结合铜离子,在铜转运体系中,CopC作为氧化还原开关参与胞内铜的调控,CopC中央的疏水桶结构及两端金属离子结合性质对CopC铜转运功能的影响尚不清楚。牛血清白蛋白(BSA)则不同,它是血浆中最丰富的载体蛋白,包含583个氨基酸残基,是富含α螺旋的球蛋白,结构较为疏松且蛋白质表面亲水性较强,能够与许多生物分子和金属离子相互作用。N-EoCen分子量与CopC相近,结构上则与BSA较为类似。
蛋白质分子结构复杂性造成其吸附的复杂性,预测CopC与BSA、N-EoCen明显不同的三维结构特征,会导致在玻碳电极表面吸附行为有显著差异,同时吸附状态蛋白质的金属结合活性是否保留也需进一步探知。由于Eu3+离子具有丰富的光、电性质,因此,本文拟选择Eu3+为探针,通过对CopC,BSA,N-EoCen的吸附性及与Eu3+离子相互作用的比较研究,为揭示吸附状态蛋白质与其生理功能的关系提供新的证据。
1 实验部分
1.1 主要仪器和试剂
N-2-羟乙基哌嗪-N-2-乙磺酸(Hepes),铁氰化钾(K3[Fe(CN)6]),牛血清白蛋白(BSA),是Sigma分装产品,未进一步纯化。湖南稀土金属材料研究所产品三氧化二铕(Eu2O3),质量百分数达99.99%。氯化钾(KCl)购于BBI公司,质量百分数>99%,其他化学试剂均为分析纯。CHI-660C电化学工作站(上海辰华仪器有限公司)为测定用电化学工作站。
1.2 实验方法
1.2.1 电化学测定方法
电解池使用三电极体系,饱和甘汞电极(SCE)为参比电极,工作电极使用玻碳电极(GC),辅助电极为铂网,实际测定的电位均相对于SCE给出。每次实验前使用 0.3 μm、0.05 μm Al2O3粉末在抛光布上将工作电极打磨成镜面,再使用超声波清洗仪于乙醇、蒸馏水中清洗后使用。制备BSA修饰电极(BSA-GC)操作过程:将玻碳电极打磨清洗干净后放入10 mmol·L-1Hepes(pH 7.4)缓冲溶液中,缓冲溶液含有100 mmol·L-1KCl,1.0 mmol·L-1的Fe(CN)63-,4.0 μmol·L-1BSA,静置100 min,由于BSA对玻碳电极的直接吸附作用将BSA固定在电极表面,制成BSA-GC电极。循环伏安实验扫描电势范围为—0.2 V~0.5 V,设置扫描速度为50 mV·s-1。实验在含有 1.0 mmol·L-1Fe(CN)63—,100 mmol·L-1KCl的10 mmol·L-1Hepes缓冲溶液中进行。
1.2.2 Eu3+储备液的配制
将称量好的Eu2O3用稀盐酸溶解,控制溶液pH约5~6的微酸性条件,用EDTA标准溶液对一定量的该溶液进行滴定(盐酸六次甲基四胺缓冲液,pH 5.7),最终给出其准确浓度。
1.2.3 蛋白储备液的制备
文中使用的N端野生型八肋游仆虫中心蛋白(NEoCen),按照文献[11]中方法诱导、表达和纯化得到。铜伴侣蛋白CopC则按照文献[12]进行诱导、表达和纯化,得到的两种蛋白质均存于10 mmol·L-1Hepes中,经十二烷基磺酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)测定,为单一条带,纯度达到实验要求。
1.2.4 蛋白浓度的测定
CopC和N-EoCen都含有酪氨酸残基,蛋白浓度可依据280 nm处所含酪氨酸残基的紫外吸收来确定。Pace等人[13]提出蛋白质的摩尔吸光系数计算方法,即:基于发光氨基酸含量的方法。按照此方法确定NEoCen的摩尔吸光系数为ε280= 4 350 L·mol-1·cm-1;CopC的ε280= 6 970 L·mol-1·cm-1。应用HP8453 UVVis吸收光谱仪,光程为1 cm的石英比色皿,25℃温度下,进行紫外吸收测定。
2 结果和讨论
2.1 CopC,N-EoCen及BSA在玻碳电极表面的吸附性
蛋白质在电极表面吸附性受溶液条件(pH,蛋白浓度,离子强度)制约同时也受电极表面性质的影响。图1是BSA、CopC和N-EoCen分别加入10 mmol·L-1Hepes(pH 7.4,100 mmol·L-1KCl)缓冲溶液中引起铁氰化钾循环伏安(CV)变化的图。图1A是加入4.0 μmol·L-1BSA连续测定的CV曲线。当BSA浓度为0时,铁氰化钾呈现为一可逆的氧化还原行为(图1A,曲线a),BSA加入后,氧化还原峰电流随时间的延长而逐渐减弱,氧化还原峰电位差则随时间的延长而逐渐增大(图1A,b-k)。BSA加入后,连续100 min的CV扫描见图1D曲线4,由(Ip,c(t)/(Ip,c(0)对时间作图所得,Ip,c(0)为0时刻的还原峰电流。由图可示,随着扫描时间的延长Ip,c在逐渐减小,说明由于BSA吸附于玻碳电极表面,Fe(CN)63-与玻碳电极之间的电子传递受阻,从而引起电流下降,氧化还原峰电流可逆性逐渐降低。当连续扫描CV约300 s时相应的峰电流下降了约47%,之后变化趋势渐缓(图2),一级吸附速率常数经拟合得0.056 9±0.004 2 s-1(见图2,表1)。可由还原峰电流减小计算得最终表面覆盖率达到56%。以CopC代替BSA进行相同的实验,在相同实验条件下加入4.0 μmol·L-1CopC连续测定CV曲线(图1C),CopC浓度为0时,铁氰化钾呈现为一可逆的氧化还原行为(图1C,曲线a),CopC加入之后,氧化还原峰电流并未随时间延长而逐渐减弱,氧化还原峰电位差也未随时间逐渐增大(图1C,b-k)。由(Ip,c(t)/(Ip,c(0)对时间作图得 1D 曲线 1。从图可见扫描到28 s时峰电流升高13%,之后在1 000 s内又回落到100%,在1 000 s~6 000 s的时间范围内还原峰电流基本维持在100%初始峰电流处,表明CopC并未在玻碳电极表面发生吸附,28 s处峰电流的升高有可能是CopC带有一定正电荷所致。在相同条件下N-EoCen的吸附实验得图1B、图1D曲线3和图2,从图可见在扫描到700 s时峰电流的变化才逐渐变缓,相应的峰电流下降了约41%,1 000 s范围内拟合得到一级吸附速率常数为(0.012 8±0.001 3)s-1(见图2,表1),表面覆盖率最终达到55%。实验表明N-EoCen也在玻碳电极表面发生吸附。为进一步证实探针离子的氧化还原峰电流减小是由于蛋白质在玻碳电极上吸附所致,进行了相应的空白实验。图1D曲线2是在相同的实验条件下,未加入任何蛋白质,1.0 mmol·L-1铁氰化钾探针离子在10 mmol·L-1Hepes缓冲溶液中连续扫描CV,Ip,c(t)/Ip,c(0)随时间变化的图。由1D曲线2可见,随时间还原峰电流仅有稍许下降,这可能是电极表面少量铁氰化钾聚集造成电极污染所致,与图1D曲线3、4的还原峰电流下降程度相比,几乎可以忽略不计。

表1 CopC、BSA和N-EoCen的吸附参数Table 1 Adsorption Parameters obtained for CopC、BSA and N-EoCen
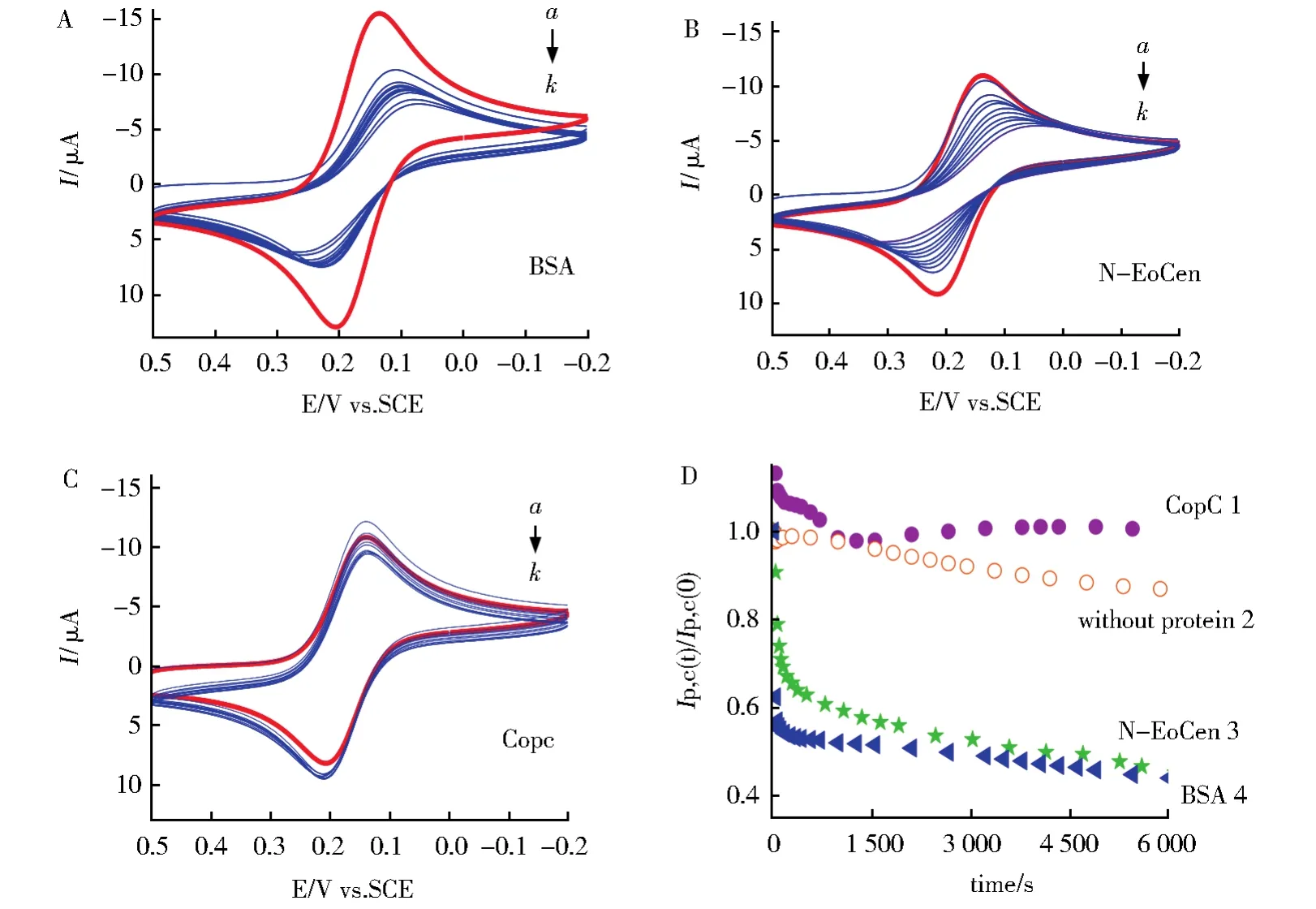
图1 CopC,BSA和N-EoCen对Fe(CN)63-离子循环伏安的影响。A:BSA加入前后Fe(CN)63-离子的CV变化图。曲线a为BSA加入之前的循环伏安图;曲线b-k为BSA(4.0 μmol·L-1)加入之后连续扫描第1,2,3,4,10,20,50,100,200,280个循环的循环伏安图。B:N-EoCen加入前后Fe(CN)63-离子的循环伏安变化图。曲线a为N-EoCen加入之前的循环伏安图;曲线b-k为NEoCen(4.0 μmol·L-1)加入后连续扫描第1,2,3,4,10,20,50,100,200,280个循环的循环伏安图。C:CopC加入前后Fe(CN)63-离子的循环伏安变化图。曲线a为CopC加入之前的循环伏安图;曲线b-k为CopC(4.0 μmol·L-1)加入之后连续扫描第1,2,3,4,10,20,50,100,200,280个循环的循环伏安图。D:加入蛋白浓度为0时(○)和加入BSA(▲)、N-EoCen(★)和CopC(●)后,Fe(CN)63-离子的还原峰电流与加入蛋白质之前初始还原峰电流之比(Ip,c(t)/Ip,c(0))随时间变化的图。实验在扫描速率为 50 mV·s-1条件下,含有Fe(CN)63—1.0 mmol·L-1,KCl 100 mmol·L-1 的 10 mmol·L-1Hepes缓冲溶液中进行Fig.1 Effect of CopC,BSA and N-EoCen on the CV of Fe(CN)63—.A:CV of Fe(CN)63—in the absence(a)and presence of 4.0 μmol·L-1BSA.The cycle numbers are1,2,3,4,10,20,50,100,200,280 from b to k;B:CV of Fe(CN)63—in the absence(a)and presence of 4.0 μmol·L-1N-EoCen.The cycle numbers are1,2,3,4,10,20,50,100,200,280 from b to k;C:CV of Fe(CN)63—in the absence(a)and presence of 4.0 μmol·L-1CopC.The cycle numbers are1,2,3,4,10,20,50,100,200,280 from b to k;D:Cathodic current profile for Fe(CN)63— in the absence(○)and presence of 4.0 μmol·L-1N-Eo-Cen(★),BSA(▲)and CopC(●).Plot of Ip,c(t)/Ip,c(0)vs.time.The data recorded in 10 mmol·L-1Hepes buffer(pH 7.4),100 mmol·L-1KCl and 1.0 mmol·L-1Fe(CN)63—,using a potential sweep rate of 50 mV·s-1
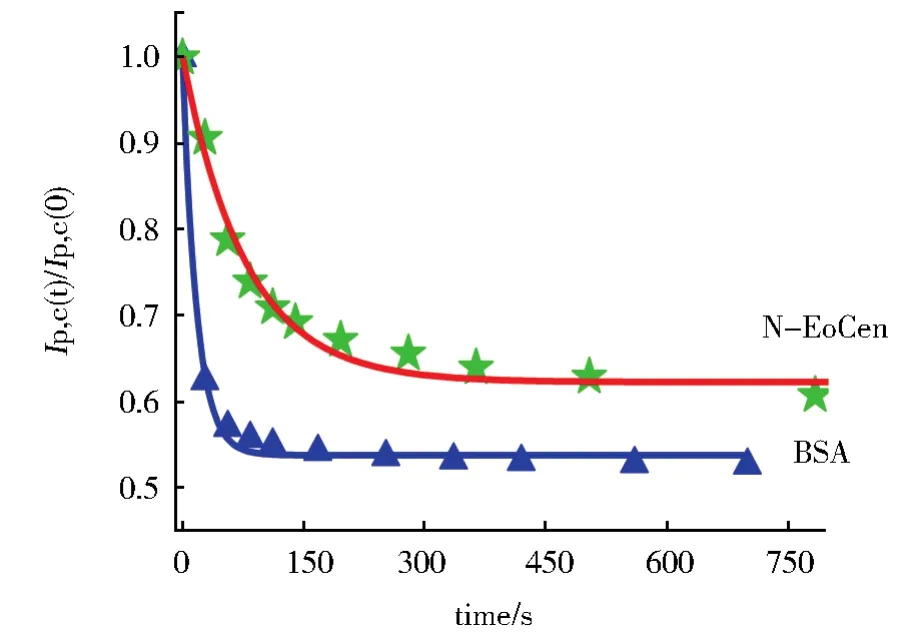
图2 加入4.0 μmol·L-1N-EoCen(★),BSA(▲)后,(Ip,c(t)/Ip,c(0))随时间变化曲线和相应的动力学拟合曲线Fig.2 Plot of Ip,c(t)/Ip,c(0)vs.time and corresponding dynamic fitting curve in the presence of 4.0 μmol·L-1N-EoCen(★)and BSA(▲)
BSA包含583个氨基酸残基,分子较大(约66 kDa),富含α-螺旋,是结构较为松散的亲水性球蛋白。N-EoCen在结构上与BSA有一定相似性,也是富含α-螺旋的结构较为疏松的亲水性球蛋白[14],不同的是分子较小(10 kDa),由101个氨基酸残基组成。CopC则与前两者都非常不同,其结构较为致密,不含α-螺旋,由102个氨基酸残基构成,分子较小且表面疏水性较强,在溶液中呈现由9股β折叠围成的桶状结构。
影响蛋白质吸附性的因素很多,蛋白质本身的内部结构稳定性是影响吸附强弱的主要因素,一般结构较疏松、分子较大的蛋白质比结构致密、分子较小的蛋白质吸附性强[5-6]。此外与电极表面性质有关,有研究显示,玻碳电极表面经过抛光打磨后会成为含较多羧基和羟基等亲水性基团的表面[15-16],因此会易于吸附具有亲水性表面的蛋白质。实验显示三种蛋白质中,BSA和N-EoCen都在玻碳电极表面表现出较强的吸附性,最终的电极覆盖率达到了55%以上,这是因为这两种蛋白质都是富含α-螺旋,结构疏松的亲水性蛋白,与亲水性的电极表面有较强的亲和作用[17]。BSA的一级吸附速率是N-EoCen的4.5倍,可能源于BSA分子较大(分子量约为N-EoCen的6倍)。在相同的实验条件下,加入CopC后扫描CV,氧化还原峰电流未减小(t=6 000 s,Ipc(t)/Ipc(0)=100%)(图1C,图1D曲线1),甚至在28 s时间范围内增大13%,抑制了在空白实验时呈现的由于铁氰化钾自身对电极污染造成的电流下降。实验充分说明CopC未在玻碳电极表面吸附,这可能一方面是由于CopC分子较小(10 kDa),是富含β折叠片的致密结构,不利于电极表面吸附,另一方面是由于CopC蛋白质表面较疏水,与亲水性玻碳电极表面不易结合。实验也同时说明,蛋白质吸附性实验能够较好地反映蛋白质的一些结构特性,例如整体结构疏松还是致密、分子量大小、α-螺旋含量,蛋白质表面的亲水性、疏水性等。
另外,静电结合作用也是影响吸附性的一个重要因素,由于BSA与N-EoCen等电点相近,(BSA为4.7,EoCen为4.8),在实验条件下(pH = 7.4),BSA和N-EoCen均带有一定的负电荷,而电极表面是荷负电的,因此推测吸附过程中蛋白质的构象变化或脱水作用引起的熵增弥补静电斥力对吸附的影响[5-6],使BSA和N-EoCen在玻碳电极表面发生吸附。CopC的等电点为8.27,因此在pH=7.4的实验条件下,CopC带有一个单位的正电荷,与荷负电表面存在静电引力作用,但实验表明CopC在玻碳电极表面不吸附,说明静电引力对吸附过程贡献较小,蛋白质吸附主要决定于蛋白质结构的内部稳定性。
2.2 以Eu3+为探针研究吸附状态BSA及N-Eo-Cen的金属离子结合活性
图3 A为使用BSA-GC电极,以Eu3+滴定,溶液中Fe(CN)63-的循环伏安随Eu3+浓度增加而逐渐变化的图。由3A所示,随着Eu3+的滴加,氧化还原峰电位差有规律减小的同时氧化还原峰电流强度亦有规律地增强,表明由于Eu3+的加入促进了Fe(CN)63-与电极之间的电子传递,说明Eu3+与电极表面的BSA发生了相互作用,这一结合作用引起BSA与玻碳电极表面的吸附强度减弱,Fe(CN)63-透过BSA膜的程度增加,因此观察到了增大的氧化还原峰电流响应。将图3A中的Ip,c/Ip,c(0)(Ip,c(0)表示Fe(CN)63-在裸玻碳电极表面的还原峰电流强度)和ΔEp分别对浓度比[Eu3+]/[BSA]作图得到图3C、3D曲线2。相同实验条件下N-EoCen与Eu3+结合实验见图3B和3C、3D曲线1。由图可见,Eu3+对BSA和N-EoCen滴定曲线影响的趋势明显不同,前者呈弧线型,具有一个拐点,后者呈S型,具有两个拐点。由于金属离子与N-EoCen结合诱导蛋白质发生显著构象改变(分子整体由“closed”向“open”状态改变[18-19])的同时发生自聚集[20],因此导致N-EoCen在电极表面的吸附力显著下降,Fe(CN)63-透过蛋白质吸附膜的程度显著增大,因此观察到了增强的电化学响应,Eu3+的结合最终使还原峰电流回升到初始还原峰电流Ip,c(0)的67%。又因为光谱方法已经证明N-EoCen具有2个稀土离子结合位点[7-8],因此电化学滴定曲线出现两个拐点,而呈明显S型上升的滴定曲线则说明Eu3+与两个位点的结合分别引起蛋白质吸附强度下降程度是不同的,暗示着Eu3+诱导的两位点蛋白质构象变化不同。应用光谱方法测定Tb3+与N-EoCen相互作用,得到的滴定曲线亦呈弧形上升[21],但仅有一个拐点,说明Tb3+与两位点结合引起的酪氨酸残基疏水性环境改变相近,因此只出现一个拐点。使用光谱方法虽能通过计算得出Tb3+与N-EoCen结合比为2∶1,但是由滴定曲线不能直接区分中心蛋白N端两个不同的金属离子结合位点。电化学滴定实验则不同,不仅能够测出结合位点数为2,同时也能通过计算得出与光谱实验相吻合的结合常数[22],更重要的是电化学滴定曲线能够更好地反映蛋白质三维构象改变。BSA是血液中一种重要的运输蛋白,分子较大,约为N-EoCen的6.6倍,与电极表面的吸附力较强,Eu3+与BSA相互作用的滴定曲线类似于脂质膜的滴定曲线[23]即呈上升弧线型。由于Eu3+的结合使还原峰电流回升到初始还原峰电流Ip,c(0)的54%。说明其结合作用引起的构象改变也类似于脂质膜,不是显著的构象变化,而是表现出均匀的表面微结构改变。实际上用Eu3+滴定BSA的过程中CD谱几乎没有变化,表明结合过程不影响BSA的空间构象,提示稀土离子有可能只结合在BSA分子表面[24],红外光谱实验则显示稀土离子可能和肽键上的C=O及边羧基上的C=O发生配位作用[25],这些报道进一步说明Eu3+与BSA的结合引起蛋白质构象改变是较为均匀的表面微结构变化,反映在电化学滴定曲线上是均匀的弧线型上升。
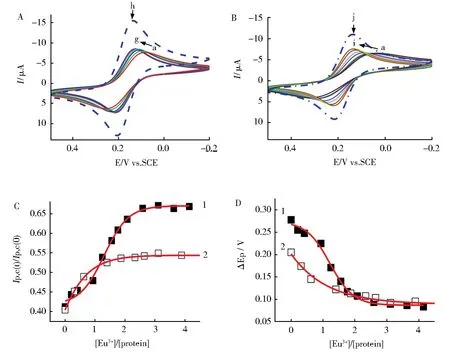
图3 BSA和N-EoCen与Eu3+相互作用的循环伏安变化图。A:BSA与Eu3+作用的循环伏安图。溶液中滴加Eu3+,加入的[Eu3+]/[BSA]分别是a→g:0,0.31,0.62,1.40,1.87,3.11,5.45。h为裸玻碳电极上探针离子的CV曲线。B:N-EoCen与Eu3+作用的循环伏安图。滴加的[Eu3+]/[N-EoCen]分别是a→i:0,0.21,0.41,0.93,1.25,1.56,1,76,2.07,2.59。j为裸玻碳电极上探针离子的CV曲线。C:(1)从B图得出的Ip,c/Ip,c(0)对[Eu3+]/[N-EoCen]作图;(2)从A图得出的Ip,c/Ip,c(0)对[Eu3+]/[BSA]作图。D:(1)从B图得出的ΔEP对[Eu3+]/[N-EoCen]作图;(2)从A图得出的ΔEP对[Eu3+]/[BSA]作图。实验在扫速为50 mV·s-1条件下,含有Fe(CN)6 3—1.0 mmol·L-1,KCl 100 mmol·L-1的10 mmol·L-1Hepes缓冲溶液中进行Fig.3 CV profile related to the interaction of Eu3+with BSA and N-EoCen.(A)The CV of the interaction between BSA and Eu3+.The concentration ratio of[Eu3+]/[BSA](from a to g):0,0.31,0.62,1.40,1.87,3.11,5.45.h is the CV of Fe(CN)63-at bare GC electrode;(B)The CV of the interaction between N-EoCen and Eu3+.The concentration ratio of[Eu3+]/[N-EoCen](from a to i):0,0.21,0.41,0.93,1.25,1.56,1,76,2.07,2.59.j is the CV of Fe(CN)63-at bare GC electrode;(C)Ip,c/Ip,c(0)(D)ΔEPas a function of[Eu3+]/[N-EoCen](1)and[Eu3+]/[BSA](2)
BSA与Eu3+反应关系式如下:
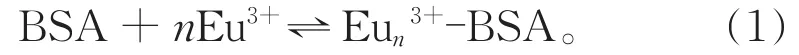
假设每一个滴定点的还原峰电流增量为△Ii,最大的还原峰电流增量为△I∞,每一个△Ii都源于Eu3+与BSA的结合,因此表达式如下:
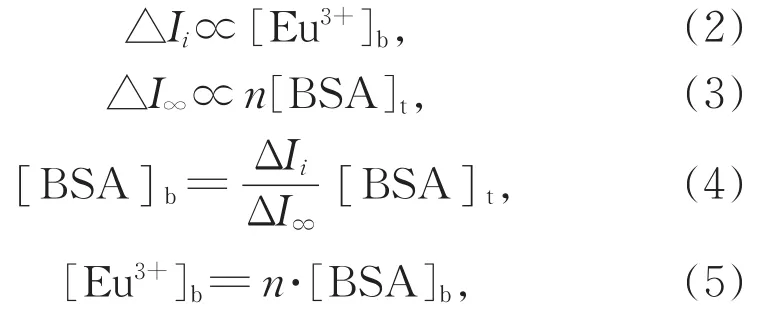
式中[Eu3+]b是结合的 Eu3+的浓度,[BSA]b和[BSA]t分别是结合了Eu3+的蛋白浓度和蛋白质总浓度。结合常数则可表示为:
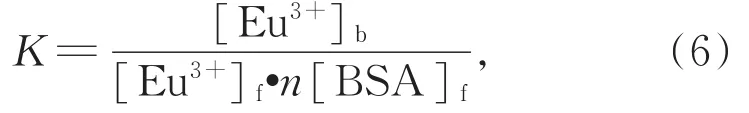
其中[Eu3+]f和[BSA]f是Eu3+和BSA的游离浓度。据以上公式可得:
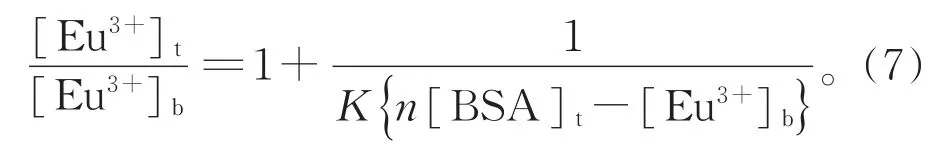
设n=1,2,3…等自然数,依据图3的滴定曲线和公式(7),以 1/(n[BSA]t-[Eu3+]b)为横坐标,以[Eu3+]t/[Eu3+]b为纵坐标作图可得图 4,满足截距尽可能接近1,线性相关系数最大的n为Eu3+离子在BSA上的结合位点数,经拟合的直线斜率倒数就是结合常数K。经计算得n=1,K=4.26×106(mol/L)-1,与文献报道的 La(III)(K=8.12×106(mol/L)-1)和 Nd(III)(K=3.85×106(mol/L)-1)结合常数数量级相近[25]。
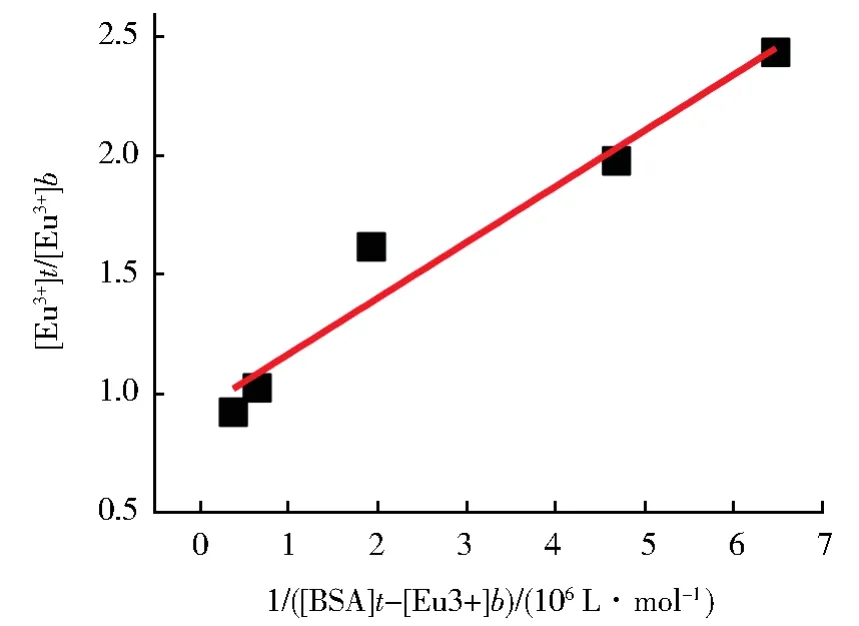
图4 [Eu3+]t/[Eu3+]b对1/(n[BSA]t-[Eu3+]b)作图Fig.4 Plot of[Eu3+]t/[Eu3+]bvs.1/(n[BSA]t-[Eu3+]b)
3 结论
利用循环伏安法研究比较CopC、BSA及NEoCen的吸附性显示,CopC在电极表面不吸附,BSA与N-EoCen均在玻碳电极表面有较强的吸附性,吸附过程静电作用贡献较小,主要决定于蛋白质结构的内部稳定性。BSA的一级吸附速率((0.056 9 ± 0.004 2)s-1)大于N-EoCen((0.012 8± 0.001 3)s-1)。利用BSA、N-EoCen的吸附性将其修饰于玻碳电极表面,以Eu3+为探针的实验显示,Eu3+对N-EoCen的滴定曲线呈S型,对应于结合过程的显著构象变化,出现2个拐点表明两个位点的结合引起的构象变化亦不同。Eu3+对BSA的滴定曲线呈上拱弧线型,暗示着Eu3+结合引起蛋白质构象改变是较为均匀的表面微结构变化。经计算Eu3+与BSA的结合位点数n=1,结合常数K=4.26×106(mol/L)-1。本研究不仅证明不同结构的蛋白质在玻碳电极上的吸附亲和力大小有很大差别,同时证明处于吸附状态蛋白质依然可以保持其与稀土离子的结合性质,并且此电化学滴定方法能够区分结合作用引起的构象变化差异。
猜你喜欢
肝博士(2022年3期)2022-06-30
汽车工程师(2021年12期)2022-01-18
中学生数理化(高中版.高考理化)(2021年10期)2021-12-06
海外星云(2021年9期)2021-10-14
高考·中(2019年6期)2019-09-10
中学生数理化·高一版(2016年7期)2016-12-07
试题与研究·中考化学(2016年1期)2016-09-30
中学化学(2016年4期)2016-05-30
中学科技(2015年8期)2015-08-08
中学生数理化·高二版(2008年1期)2008-10-19
