
融合基因与膀胱癌
2021-03-03 04:25肖俊文苏港林刘宇辰
医学新知 2021年1期
孙 浩,肖俊文,胡 坤,苏港林,刘宇辰
1. 深圳大学第一附属医院/深圳市第二人民医院泌尿外科(广东深圳 518000)
2. 深圳市泌尿生殖系统肿瘤研究重点实验室(广东深圳 518000)
3. 安徽医科大学深圳市第二人民医院临床医学院泌尿外科(广东深圳 518000)
4. 汕头大学深圳市第二人民医院临床医学院泌尿外科(广东深圳 518000)
5. 深圳大学第一附属医院/深圳市第二人民医院转化医学研究院(广东深圳 518000)
融合基因自从被科学家们发现以来,就一直被当作可能是影响肿瘤进展的重要因素。科学家针对融合基因的研究从未中断,取得了许多突破性的成就,尤其是针对BCR/ABL 融合基因的研究,开发出的伊马替尼可用于治疗慢性髓系白血病(chronic myeloidleukemia, CML)[1-2],成功挽救了无数患者的生命。近年来,随着测序技术的不断发展,越来越多的融合基因在其他的肿瘤中也被发现[3]。
1 融合基因的研究概述
1.1 融合基因的发现
第一个融合基因在20世纪70年代发现,大约在此后的十年间染色体重排是通过显带方式识别的。虽然以前已经发现重复的结构重排,但是直到20世纪70年代染色体显带技术的出现,肿瘤细胞的基因组才能被详细分析,主要的例子就是CML中的费城染色体[4]。1973年第一次报道在CML中发现的费城染色体被证明是9号染色体和22号染色体t(9;22)(q34;q11)之间移位所产生的衍生物,同时在急性髓系白血病中发现t(8;21)(q22;q22)[5-6]。这些发现激发了人们对于血液系统肿瘤的兴趣,在其中发现越来越多的染色体重排,包括在Burkitt淋巴瘤中发现的t(8;14)(q24;q32)、t(2;8)(q24;q32)和 t(8;22)(q24;q11)[7-8],在急性淋巴细胞白血病(ALL)中发现的t(4;11)(q21;q23)[9],在急性早幼粒型白血病(APL)中发现的t(15;17)(q22;q21)[10]和在滤泡细胞淋巴瘤中发现的t(14;18)(q32;q21)等[11]。在血液系统肿瘤中融合基因被大量发现后的十年中,在恶性实体肿瘤中也发现了大量的融合基因。在实体肿瘤内新发现的融合基因与在血液系统肿瘤中发现的融合基因一样具有明显的特异性,例如在肺泡横纹肌肉瘤中发现的t(2;13)(q36;q14)[12],在尤因式肉瘤中发现t(11;22)(q24;q12)[13],在肾癌中发现的 t(X;1)(p11;q21)[14],在唾液腺囊性癌中发现的 t(6;9)(q23;p23)[15]。此外许多良性的肿瘤也存在明显的融合基因,例如在脂肪瘤中发现的 t(3;12)(q27;q13)[16]。
1.2 融合基因检测的升级
1.2.1 染色体显带技术与高通量测序
传统的检测方法主要是染色体显带技术,尽管这种技术有很多优势(曾经发现过很多融合基因),但其依然存在一些十分明显的缺陷。20世纪90年代开始兴起的基因分析高通量测序工具给融合基因的检测带来了全新的革命,如阵列的基因表达平台和拷贝数分析,提供了新的途径检测融合基因。基于阵列的平台不但能提供全基因组的视图,还能提供比染色体显带更高的分辨率,并且不需要预先培养细胞。基于此技术发现的第一个融合基因是在横纹肌肉瘤中发现的PAX3-NCOA1融合基因[17],随后不久在前列腺癌中也发现了复发基因融合。Tomlins等发现了TMPRSS2基因与两个编码ETS转录因子的基因,一个是ERG导致了TMPRSS2-ERG的融合,另一个ETV1导致了TMPRSS2-ETV1的融合[18]。重要的是有一些像TMPRSS2-ERG的融合,两个基因是位于同一个染色体条带之上,无法进行染色体显带分析,这是第一次发现在常见的恶性肿瘤中存在此种基因融合的方式。随后,科学家们试图通过关注单个外显子而不是整个基因组来识别基因的5’和3’末端的差异表达,之后被证明可以成功的检测到许多其他肿瘤类型的基因融合,如腱鞘巨细胞肿瘤、肺癌和软骨肉瘤[19-21]。虽然平衡易位是基因融合最常见的方式,但是也存在着非平衡易位的方式,例如间质缺失。这种融合的例子在20世纪90年代就已经被发现,例如T细胞ALL中的STIL-TAL1融合[22]。此外有许多看似平衡的易位导致的基因融合都或多或少存在广泛的缺失、重复和扩增的断点区域[23-25]。因此,受拷贝数变化影响的基因是参与融合的潜在候选基因。以此种检测方式检测到的第一个融合基因的例子是在CML中发现的USP16-RUNX1[26]和在T细胞ALL中发现的SET-NUP214[27],随后在其他血液系统恶性肿瘤和实体肿瘤中也发现了同样的融合[28-30]。然而,这项技术并没有被当作检测基因融合的独立技术,制约它发展的主要原因是它既受恶性肿瘤典型的断点数量的影响,同时也受其他很多影响拷贝数的因素影响。
1.2.2 深度测序技术
与上述基因融合检测的指导方法不同,在十年前一种叫做深度测序技术的方法给人们提供了一种全新的思路,这种检测技术可以识别在DNA或RNA水平上的融合。通过全基因组和全转录组测序的信息,可以识别那些更加隐蔽和微小的染色体改变和基因融合。首次利用深度测序技术检测肿瘤相关的融合基因研究是在已经建立的细胞系上进行的[31],随后对像乳腺癌、结肠癌、前列腺癌、肺癌和子宫内膜癌等常见癌症的初步样本进行了大量检测[32-34]。表1[35]显示了目前按照肿瘤亚型分布的所有已知融合基因数(9519个)和重复出现的基因数(373个)。其中血液系统肿瘤中的融合基因数为932个,重复出现191个;实体中融合基因数为8 512个,重复出现168个。可以看出,目前发现的融合基因总数已经接近一万个,其中90%以上都是过去5年通过各种深度测序方法鉴定的。通过深度测序检测到的融合基因在某种程度上不同于传统方法检测到的融合基因。首先深度测序能够检测到重排产生的融合,而不是通过染色体显带分析检测到的染色体段的交换,因此分辨率就要高出很多。其次染色体内部细微的重排产生的融合现在也可以被检测到,例如在单发的纤维腺瘤中染色体12的NAB2与STAT6的翻转融合和在大肠癌染色体带10q25中500kb缺失引起的VTI1A-TCF7L2的融合[34,36-38]。这种新技术加快了新融合基因的发现速度同时其成本大幅下降,大量新的融合基因在实体肿瘤特别是在膀胱癌中被发现,这为我们进一步研究和阐释融合基因在膀胱癌进展中扮演怎样的角色打下了坚实的基础。
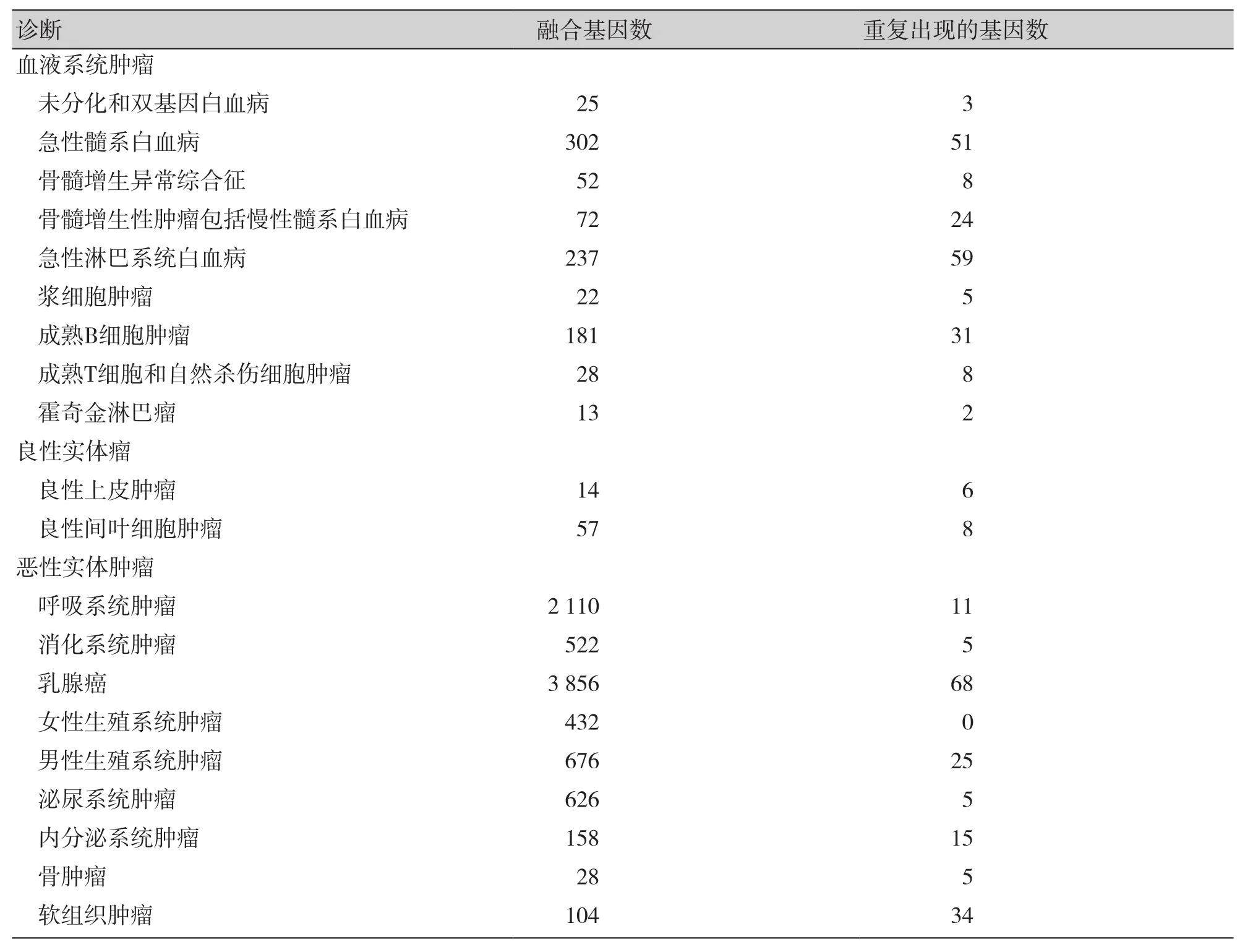
表1 参与重大肿瘤亚型的融合基因数目Table 1. Number of fusion genes involved in major tumor subtypes
1.3 融合基因的致病性
1.3.1 失去对转录的调节作用
20世纪80年代开始科学家们对各种生物当中转座子的研究日益加深,根据这些研究有科学家推测人类癌症也可能是遗传转座的结果,染色体易位和其他染色体重排导致位于断点的正常细胞基因的异常表达[39-40]。这一假设马上就被证明是正确的,第一次是在MPC和Burkitt淋巴瘤中发 现 t(2;8)(p11;q24)、t(8;14)(q24;q32)和t(8;22)(q24;q11)引起免疫球蛋白基因位点非正常重组,最终导致转录因子MYC表达下调[41]。随后,IGH、IGK和IGL易位的克隆成为识别肿瘤相关基因的一种新手段,可以用于检测许多肿瘤细胞[42-44]。在恶性T细胞疾病中发现了类似的情况,其中涉及T细胞受体基因位点的非正常重排导致了染色体断点区域附近失去了原有的调控作用[45-47]。在大多数情况下,这种重排所解除的基因大多编码转录因子或参与转录调控的其他蛋白质。众所周知转录调控的中断无疑是肿瘤发生的重要因素,然而参与其中的许多基因被重新排列这势必会导致细胞出现不正常的增殖。由于易位和其他染色体重排而导致的基因表达异常并不是B细胞和T细胞恶性肿瘤所独有的,有一些其他的实体肿瘤也是由于目标基因调控序列受到影响而被解除管制。
1.3.2 嵌合基因
20世纪80年代大量的染色体易位导致的基因融合被发现,使人们认为基因解除管制可能是染色体重排的唯一途径。因此当9q34上的ABL1基因在CML中被转移到22q11时,最初推测费城染色体的功能可能是通过22q11处来自IGL基因的启动子或增强子来使ABL1的表达增加。然而事实却正好相反,t(9;22)被证明是一个嵌合的BCR-ABL1基因,编码一个具有异常酪氨酸激酶活性的杂交蛋白[1-2],并很快就发现了几种产生融合蛋白的易位,像在ALL中发现的t(1;19)(q23;p13)和TCF3-PBX1[48-49]。从20世纪90年代开始许多嵌合基因在实体肿瘤和间充质来源的肿瘤中也被发现。起初参与融合的基因类型主要是编码转录因子或相关的辅助因子的基因,例如RARA、RUNX1和TCF3等[50]。因此,基因嵌合主要被当作是增加或抑制转录或构成激活激酶信号。当全基因组以及转录组测序成为更常用的研究和临床诊断手段时,将会有更多的嵌合基因被发现。
1.3.3 基因截断
近几年基因截断成了新的致病因素,基因截断将会导致肿瘤抑制因子的失活,例如CDKN2A[51]和NF1[52];或者导致其变为野生型蛋白有主导作用的亚型,例如PAX5[53]。现在详细染色体分析已经证明,如果这些基因被放松调节或者结构发生改变就很大程度上会发生癌变。已经有很多学者证实过基因融合会比单个的基因突变对细胞的影响更大,因为许多基因都已经被证明是通过其他基因的改变才引发其突变。例如除了在某些恶性肿瘤中易位外,有些基因在相同或其他肿瘤中也经常发生突变、删除和扩增,像BRAF、CCND1、CCND2、CEBPA和ETV6等[54-56]。
综上所述,融合基因的研究目前所取得的主要成果大多来自血液系统肿瘤,各种应用也主要集中在白血病和淋巴瘤。但是,近些年的研究也不断的证明基因融合是所有肿瘤细胞的共同特征,大家一致认为融合基因在肿瘤的进展过程中起到了关键的作用。越来越多的融合基因在不同的实体肿瘤细胞中被发现,特别是在膀胱癌中有一些已经被明确证实是肿瘤进展的关键因素。但是,也有许多融合基因我们目前还没有研究清楚它们与肿瘤的具体关系。这就需要全世界的科学家和临床医生们去不断地探索,揭示融合基因和肿瘤之间的具体联系。
2 融合基因研究在膀胱癌中的应用现状
膀胱癌是泌尿生殖系统最常见的恶性肿瘤之一,据报道在全球范围内其发病率居全部恶性肿瘤的第九位,死亡率居全部恶性肿瘤的第十三位,世界卫生组织推测膀胱癌的发病人数和死亡人数在不久的将来将会翻倍[57]。目前,关于膀胱癌的诊断、治疗和预后方面还存在许多问题亟待解决,科学家们通过深度测序发现在膀胱癌中存在大量的融合基因,但是这些融合基因在膀胱癌中的作用有很多还不明晰。被认为与膀胱癌关系最为密切,也是研究最多的是人成纤维细胞生长因子受体(FGFR)家族。
2.1 FGFR家族在膀胱癌研究中的应用
FGFR家族由22个因子和5个跨膜受体组成。在这22个因子中18个是分泌的,其中4个是完全在细胞内发挥作用的。FGFR基因改变发生在各种癌症中,包括膀胱癌、乳腺癌、卵巢癌、子宫内膜癌、肺癌和胃癌[58]。目前FGFR1/2/3/4均在膀胱癌的研究中有过报道,FGFR1的改变包括FGFR1扩增和细胞外D2结构域附近的T141R突变[59]。 FGFR基因扩增通常与基因产物的过表达导致酶活性的增加有关,例如FGFR1-NTM 的融合就是通过这种方式产生作用[59]。膀胱癌中也会发生FGFR3的扩增。此外,有5个FGFR3的细胞外结构域突变被发现,分别是:S131L、R248C、S249C、G370C 和 Y373C[60]。
很多FGFR的改变被猜测可能与膀胱癌是否发生肌层浸润有密切的关系,有报道称超过一半的非肌层浸润性膀胱癌和大约10%的肌层浸润性膀胱癌发生FGFR3突变[61]。通过重排的方式产生融合蛋白是FGFR影响肿瘤进展的常见方式,其中最常见也是研究最多的融合是FGFR3-TACC3。首次发现FGFR3-TACC3融合是在神经胶质细胞瘤中发现[62-64]。Williams等发现在膀胱癌中同样存在此种融合基因,这种融合会导致它们缺发磷脂酶Cγ1(PLCγ1)在内的最后一个外显子结合位点,从而可能导致癌变。同时,他们还指出由于FGFR3融合的尿路上皮细胞对FGFR选择型药物极为敏感,融合基因的表达可能有助于选择适合FGFR靶向治疗的患者[65]。Guo等对膀胱癌标本进行全基因组和全外显子组的测序也发现了FGFR3-TACC3的融合,并且认为FGFR3-TACC3可以作为膀胱癌诊断的潜在指标[66]。除了常见的FGFR3-TACC3融合外,还有一些融合也被报道过,例如FGFR3-JAKMIP、FGFR3-TNIP2和 FGFR3-ADD1等[61]。FGFR融合蛋白通常导致形成受体二聚体和随后的蛋白激酶磷酸化和接下来的激活来发挥作用。目前,针对FGFR家族在膀胱癌中的研究还有很多。美国FDA在2019年第一个批准了口服FGFR拮抗剂:厄达替尼(Erdafitinib)来治疗晚期的膀胱癌,并取得了很好的临床效果,这无疑是FGFR家族在膀胱癌研究中的一次重大突破。
此外,除了FGFR家族外,许多FGF基因的突变也在膀胱癌中被大量报道,其中涉及最多的是FGF3/4/19,大约在12%的病例中可以发现FGF基因突变[66]。在膀胱癌中,针对FGF3/4/19基因改变的主要方式是扩增,基因的扩增导致这些生长因子信号大量增加。 相反,膀胱癌中涉及FGF17和FGF20的基因改变大多是缺失,从而导致生长因子信号减少[61]。因此我们可以推测许多FGF基因表达的上调或下调与膀胱癌的发生发展有着重要的关系。
2.2 其他融合基因在膀胱癌中的应用
近年来RNA测序技术的兴起给了我们一个寻找融合基因的全新视角,越来越多新的融合基因通过RNA测序技术被检测出来。Helsten等通过严格筛选条件从TCGA数据库中的 819 个融合基因中筛选出 SEPT9/CYHR、IGF1R/TTC23、SYT8/TNNNI2和 CASZ1/DFFA 四个新的融合基因,但是 其 中 SEPT9/CYHR、IGF1R/TTC23 和 CASZ1/DFFA并没有在患者采集的样本中检测到,这些融合基因可能并不是膀胱癌发生的主要原因,同时他们发现SYT8/TNNNI2融合基因却存在于37.5%(18/48)的样本之中,具体的功能和临床相关性尚不清楚[67]。Kekeeva等从414个膀胱癌样本中发现了19 547个高置信度的融合基因,经过严格的筛选后得到了271个融合基因,其中有13个融合基因是首次发现。他们选取了6个融合基因用于细胞系和临床样本的实验,发现其中两个嵌合RNA BCL2L2-PABPN1和CHFR-GOLGA3在膀胱样本中的表达明显高于临近的正常样本,亲本基因的野生型没有差异表达。接着,他们证明了这两种融合是由相邻基因之间的顺式剪接产生的,而且这两种融合主要是在细胞核内被检测到,这说明具有潜在非编码RNA的作用,这些嵌合RNA或许是全新的潜在的新型生物标志物[68]。
除了通过传统的测序来发现和验证融合基因外,利用融合基因与其他技术之间的联系来治疗膀胱癌也是一个值得探索的方向。其中一些研究已经取得了一定的成绩,例如联合自杀基因通过改造和修饰腺病毒来治疗膀胱癌。自杀基因,是指将某些病毒或细菌的基因导入靶细胞中,其表达的酶可催化无毒的药物前体转变为细胞毒物质,从而导致携带该基因的受体细胞被杀死。常用的自杀基因系统有两种,TK-GCV系统和CD-5-FC系统。更昔洛韦(GCV)是临床上用于治疗单纯疱疹的药物。GCV在胸苷激酶的作用下生成三磷酸GCV,能阻断DNA合成产生细胞毒作用。病毒、细菌、真核细胞中都存在胸苷激酶(HSVTK)。Wang等构建了一种Arg-Gly-Asp(RGD)修饰的腺病毒RGDAdUPII-TK,它携带HSV-TK自杀基因,与GCV联合注射处理膀胱癌细胞系,证实了RGDAd-UPII-TK与GCV注射相结合可以显著降低T24肿瘤的生长,增加体内凋亡[69]。胞嘧啶脱氨酶基因(CD)存在于许多细菌和真菌中,5-氟胞嘧啶在微生物体内被CD代谢 成5-氟尿嘧啶,后者有明显的抗肿瘤活性。目前有学者将CD-TK两个自杀基因融合到一起形成双自杀基因,用以治疗膀胱癌。Hu等采用薄Flm水化超声、碳二酰亚胺化学方法和静电吸附方法,制备了负载CD-TK双自杀基因的均匀纳米尺度纳米气泡,并将其与可以和VEGFR2阳性细胞相结合的抗VEGFR2相融合。结果证明CD-TK双自杀基因的过度表达可以在体外培养中抑制肿瘤细胞的生长[70]。这为我们治疗膀胱癌提供了一个新的思路。
3 结语
目前全世界的科学家都认识到融合基因在肿瘤的进展过程中起了至关重要的作用。随着测序技术的不断进步,越来越多的融合基因被我们发现,相应的也有越来越多的融合基因被证明在肿瘤的进展过程中扮演重要的角色。但是,测序技术的进步对于我们研究融合基因来说是一把“双刃剑”。一方面,测序技术的不断进步使我们可以对全基因组、全转录组进行测序,而不再像以前一样依赖简单的染色体显带技术观察基因融合,这使得我们可以发现许多原来观察不到的融合基因。但另一方面,先进的测序技术可以观察到非常细微的基因融合,使我们很难分辨出哪些在肿瘤进展过程中真正发挥重要作用,哪些是无意义的融合,因为现在有研究表明我们可能夸大了某些融合基因的作用。
未来的研究中,探寻这些融合基因在肿瘤进展过程中具体扮演怎样的角色,与肿瘤的恶性程度是否相关或许才是我们应该研究的重点。伴随着合成生物学的发展与进步,我们可以利用合成生物学基于对CRIPRS-cas9的改造,使某些融合基因在特定的肿瘤组织过表达或敲除某些基因,从而确定融合基因是否真的在肿瘤的进展过程中起关键性的作用。
猜你喜欢
中国人兽共患病学报(2022年9期)2022-10-19
云南医药(2022年4期)2022-09-01
牡丹江医学院学报(2022年2期)2022-06-01
家庭医药(2022年5期)2022-05-18
中国典型病例大全(2022年7期)2022-04-22
昆明医科大学学报(2022年1期)2022-02-28
科学导报(2021年29期)2021-06-03
科海故事博览·下旬刊(2019年6期)2019-04-16
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
