
环氧合酶-2抑制剂的研究进展
2021-02-28 03:40王慧哲胡晓龙中国药科大学中药学院南京210009
西北药学杂志 2021年6期
王慧哲,胡晓龙,汪 豪(中国药科大学中药学院,南京 210009)
炎症通常是指由感染、身体伤害、心理压力或组织对抗体攻击的反应引起的细胞损伤的反映,与关节炎、癌症、抑郁症、阿尔兹海默症(AD)、帕金森氏综合征(PD)和糖尿病等疾病的发展密切相关[1-2]。环氧合酶(COX)是催化花生四烯酸代谢和前列腺素(PG)合成的初始步骤,是炎症的主要介质。
环氧合酶分为环氧合酶-1(COX-1)和环氧合酶-2(COX-2)2种同工型。COX-1在大多数组织中均能表达,被认为是体内前列腺素合成的同工型,而COX-2的表达主要受炎症诱导。COX-2的激活和随后的前列腺素E2(PGE2)的产生引起促炎细胞因子白细胞介素-1β(IL-1β)、肿瘤坏死因子-α(TNF-α)和干扰素-γ(IFN-γ)的释放[3],选择性COX-2抑制剂能抑制促炎细胞因子的释放,进而治疗炎症引起的相关疾病。其作用机制见图1。本文介绍了COX-2抑制剂与一些炎症相关疾病的作用机制,综述了近年来具有不同化学结构COX-2抑制剂的研究进展并探讨其结构活性关系,为药物的设计提供理论依据。

图1 COX-2抑制剂的作用机制Fig.1 Mechanism of COX-2 inhibitor
1 炎性疾病与COX-2抑制剂
1.1癫痫 癫痫是一种常见的多因素神经疾病,越来越多的证据揭示了神经炎症在癫痫中的病理、生理作用[4]。大脑和血脑屏障中关键炎症介质的表达增加可能影响神经元的功能和兴奋性,进而增加癫痫发作的易感性。在癫痫发作期间,COX-2激活PG信号通路,从而引发大脑的继发性损伤并加剧疾病的严重程度[5]。COX-2被认为是治疗癫痫的潜在靶标。相关研究表明,COX-2抑制剂通过减少PGE2的产生而发挥抗惊厥活性,减少钙离子的流入和释放兴奋性神经递质谷氨酸,从而阻止癫痫发作[6]。同时,COX-2抑制剂可以抑制促炎细胞因子的产生,减轻炎症反应。这种通过抑制COX-2表达的治疗方法取决于治疗剂量、给药时间、治疗持续时间和COX-2抑制剂的选择性。
1.2癌症 核因子-κB(NF-κB)与COX-2的表达存在正相关,有利于肿瘤发展的信号传导[7]。一种新型的COX-2抑制剂JTE-522通过诱导活性氧的产生抑制NF-κB的活性,最终导致人胃癌AGS细胞凋亡。此外,COX-2抑制剂不仅可以通过干扰细胞周期蛋白依赖性激酶4/6(CDK 4/6)复合物的活性阻止G1期的进展,还能影响肿瘤坏死因子(TNF)相关的凋亡诱导配体(TRAIL)受体的活性诱导肿瘤细胞凋亡[8]。因此,选择性COX-2抑制剂被认为是治疗癌症的方法之一。然而,最近研究发现,选择性COX-2抑制剂作为抗癌和抗炎药对不同组织(如肝、肾和心血管系统)可能产生多种不良影响,其潜在机制是源自COX-1的血栓烷A2(TXA2)与源自COX-2的血管保护因子前列环素(PGI2)之间的失衡[9-10]。通过使用纳米球、纳米凝胶、微粒、微乳剂以及脂质体等新型的药物递送系统可以减少这些不良反应。
1.3神经精神疾病 越来越多的证据支持神经精神疾病(如抑郁症、双相情感障碍、精神分裂症和强迫症)与炎症之间的关系,炎症通过对神经元的增殖、存活和分化产生负面影响,从而成为神经精神症状的驱动者[11]。促炎性标志物已被证明与神经精神疾病的发展相关,白细胞介素-6(IL-6)激活了肝脏中C反应蛋白(CRP)的释放,而CRP被认为是与多种神经精神疾病密切关联的蛋白之一[12]。神经递质的失调一直是神经精神疾病的焦点,促炎性细胞因子白细胞介素-2(IL-2)和干扰素-α(IFN-α)已显示出可直接增加犬尿氨酸途径的吲哚胺-吡咯-2,3-二加氧酶的活性,从而促进色氨酸向犬尿氨酸转化,该代谢物已被证明是抑郁和焦虑症状的诱因[13]。抑制COX-2的表达可减轻这种炎症负荷,从而减少这些途径对大脑的影响。因此,COX-2抑制剂成为预防和治疗神经精神疾病的新方法。
1.4关节炎和神经退行性疾病 非甾体类抗炎药(NSAIDs)抑制COX的表达,COX-2主要在炎症细胞中表达,并在慢性和急性炎症中明显上调,成为炎性疾病的关键靶标。COX-2抑制剂可在骨关节炎、类风湿性关节炎和急性疼痛等疾病中发挥抗炎、解热和镇痛作用[14]。长期以来,神经炎症与神经退行性疾病如AD和PD的发病机制有关[15],神经元和小胶质细胞在炎症期间明显上调了COX-2的表达。此外,PGE2介导的神经炎性介质TNF-α、IL-1β和活性氧(ROS)以及亚硝酸盐的释放可能导致神经元死亡[16]。
2 COX-2抑制剂的研究现状
经典的NSAIDs具有作为常规结构特征的羧酸基团,羧酸部分与精氨酸和酪氨酸发生氢键和/或离子相互作用。COX-1和COX-2抑制剂作为典型的NSAIDs,常常导致胃肠道疾病和出血等不良反应。目前,针对新型NSAIDs的研究集中在选择性COX-2抑制剂上,该抑制剂可显示出更高的耐受性。广泛的结构活性研究(SAR)确定了选择性COX-2抑制剂的主要特征:中心环必须被2个苯基围绕,并且其中1个苯环具有4-磺酰胺基或4-甲基磺酰基基团。在这些结合方式中,磺酰胺或甲基磺酰基结合在COX-2特定的侧袋中,并通过氢键稳定,研究最多的是被五元环、六元环取代的二芳基化合物以及一些天然来源的选择性COX-2抑制剂[17]。其中化学合成的COX-2抑制剂主要包括吡唑类、噁唑类、吡咯类、吲哚类、噻唑类、吡啶类和咪唑类,而天然来源的COX-2抑制剂主要为生物碱类、香豆素类和黄酮类化合物。
2.1吡唑类 吡唑是制备新型COX-2抑制剂的重要骨架。Gedawy E M等[18]合成的吡唑磺酰胺衍生物具有COX-2 和5-脂氧合酶(5-LOX)抑制作用,其中苯并噻吩-2-基吡唑羧酸类化合物 (化合物1,见图2)的镇痛和抗炎活性高于塞来昔布,其半数抑制浓度(IC50)为0.01 μmol·L-1,而且与COX-1相比具有更高的选择性(SI=44.56)。体内抗炎实验发现,给药1 h后,大鼠足肿胀得到了有效缓解,并在第2小时达到最大肿胀抑制率。Murahari M等[19]合成1,4-二氢吡喃并吡唑类化合物(化合物2,见图2),COX-1、COX-2体外抗炎试验和角叉菜胶诱导的大鼠足水肿模型实验均表明该化合物具有显著抗炎活性。当苯环上C-4位的供电子基团(-OCH3)换成吸电子基团(-F)时,化合物3(见图2)的抗炎活性有所减弱,提示苯环C-4位的供电子基团对此类化合物的活性有重要作用。Abdellatif K R A等[20]报告了二芳基吡唑衍生物(化合物4,见图2),该化合物在中心五元杂环上连接邻位二芳基环,其中一个芳基环上的甲磺酰基(-SO2CH3)具有COX-2选择性,另一个芳基环对位连接不同取代基得到的化合物活性由高到低依次为4-SO2NH2>4-COOH>4-H>4-SO2CH3。大鼠体内抗炎实验表明,化合物4能够有效抑制角叉菜胶诱导的大鼠足水肿。该课题组设计的1,3,4-三取代-吡唑衍生物(化合物5,见图2)与塞来昔布相比,具有显著COX-2抑制作用和选择性,体内抗炎实验表明,该化合物具有较优的抗炎活性,给药1 h即能有效抑制大鼠足肿胀,且在给药的第5小时依然保持较高的足肿胀抑制作用。此外,化合物5与布洛芬相比具有更低的致溃疡性。分子对接表明,该化合物与塞来昔布具有相似的结合模式和相互作用,而且具备形成更多氢键的特征[21]。
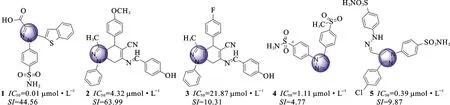
图2 化合物1~5的结构式Fig.2 Structures of compounds 1-5
2.2噁唑类 Rakesh K S等[22]合成了异噁唑衍生物。体外抗炎试验显示,化合物6(见图3)表现出与吲哚美辛相当的COX-2抑制作用。在计算机模拟研究中,该化合物与吲哚美辛相比,具有更高的原子接触能,能更牢固地与COX-2的催化域结合。此外,其COX-2抑制作用可能是由具有强供电子取代基的芳基和中等电子富集的杂芳基取代基(如吲哚)引起的。Kaur A等[23]合成了N-(3,4-二甲氧基苯基)-苯并噁唑类化合物 (化合物7~9,见图3),体外抗炎试验表明,化合物7比塞来昔布和布洛芬具有更好的COX-2抑制作用和选择性,化合物8和9显示出较好的COX-2抑制活性。角叉菜胶诱导的大鼠足水肿实验发现,化合物7~9显示出比布洛芬更明显抑制大鼠足肿胀的作用,并且该化合物在胃黏膜上显示出良好的安全阈度。对接分数表明,苯环邻位上不同吸电子基团取代化合物的抗炎活性由高至低依次为:2-Cl>4-NO2>2-Cl,4-NO2。

图3 化合物6~9的结构式Fig.3 Structures of compounds 6-9
2.3吡咯类 Kim K J等[24]合成的化合物10~12(见图4)被认为是比塞来昔布更有效和更具选择性的COX-2抑制剂。对接结果显示,化合物10在COX-2结合位点内呈现有利的取向,并且以与塞来昔布几乎相同的方式结合到COX-2的活性位点,化合物10的-SO2NH2部分的2个氧原子与精氨酸和苯丙氨酸表现出2个氢键结合,-NH2上的2个氢原子分别与异亮氨酸和谷氨酰胺形成2个氢键。见图5。此外,苯环对位取代基的不同导致化合物活性有较大差异,其活性顺序由高到低依次为:-Cl>-H>-OCH3,吸电子基团取代比供电子基团取代的化合物具有更强的COX-2抑制活性。Reale A等[25]合成了一系列新的1,5-二芳基吡咯-3-硫衍生物(化合物13~14,见图4),化合物13和14在体外抗炎试验中均表现出明显的COX-2抑制活性。角叉菜胶诱导的大鼠足肿胀实验表明,化合物13~14给药30 min后的肿胀抑制率分别为95.39%、94.97%。在大鼠腹部收缩实验中,以20 mg·kg-1的剂量进行灌胃,2种化合物均能显著减少大鼠的扭体次数。

图4 化合物10~14的结构式Fig.4 Structures of compounds 10-14
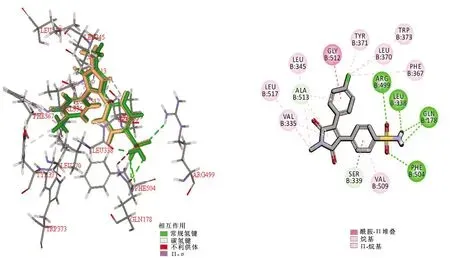
图5 化合物10与COX-2的结合图注:橙色:化合物10;绿色:塞来昔布;蛋白库编号(PDB ID):3LN1。Fig.5 Binding of compound 10 with COX-2Notes:orange:compound 10;green:celecoxib;protein library number (PDB ID):3LN1.
2.4吲哚类 Singh P等[26]合成了N-1位具有甲苯-4-磺酰基、C-3位具有二肽基团的吲哚类化合物15(见图6)。该化合物不仅具有比双氯芬酸钠更明显的COX-2抑制作用,还在体内抗炎实验中表现出良好的抗炎活性。二肽基团的存在、C-3取代基上氨基酸的组合以及N原子上的甲苯磺酰基是增加COX-2抑制作用和抗炎活性的原因。此外,分子对接结果表明,氨基酸C-α位的S构型增加了与COX-2活性位点的相互作用。Laube M等[27]设计了吡咯并[3,2,1-hi]吲哚类化合物(化合物16~18,见图6),化合物16~18是具有与塞来昔布相似或更高亲和力的COX-2抑制剂。其取代基的大小直接影响COX-2抑制作用的强弱,氯原子取代氟原子使化合物17的COX-2抑制作用提高了10倍,此外,将甲基磺酰基取代基从4-苯基环转移到5-苯基环也可以提高化合物的体外抗炎活性。Lamie P F等[28]合成了N-取代的吲哚衍生物(化合物19~21,见图6),体外COX-2抑制研究发现,化合物19~21与塞来昔布相比具有显著抗炎活性和合理的选择性指数,其结构的共同特点是苯甲酰基部分取代了吲哚的-NH基团。在预处理的人脐静脉内皮细胞中使用酶联免疫吸附试验(ELISA)测试化合物19~21的体外抗炎特性,给药浓度为50 μmol·L-1时能够有效抑制NF-κB的表达。

图6 化合物15~21的结构式Fig.6 Structures of compounds 15-21
2.5噻唑类 噻唑和噻唑烷酮结构不仅存在于多种天然来源的化合物中,还被广泛应用于化学合成的药物中[29]。El-Achkar G A等[30]设计并合成了2种新型的噻唑衍生物(化合物22和23,见图7),二者对COX-2依赖性PGE2的产生均具有显著抑制作用。化合物22是一种非选择性COX-1/COX-2抑制剂,而化合物23具有选择性COX-2抑制作用。此外,在大鼠气囊炎症模型中,化合物22和23均显示出良好的抗炎活性。Abdellatif K R等[31]设计的噻唑烷-4-酮衍生物(化合物24和25,见图7)表现出非常接近塞来昔布的COX-2抑制能力和选择性指数。体内抗炎实验表明,化合物24和25给药3 h后,比参比药物塞来昔布具有更高的抗炎活性。进一步实验研究发现,化合物24的半数有效量(ED)50=27.7 μmol·kg-1,与塞来昔布(ED50=28.2 μmol·kg-1)的口服抗炎活性几乎相当,而化合物25(ED50=18.1 μmol·kg-1)比塞来昔布更有效。致溃疡实验进一步证实了化合物24和25具有较高的安全性。Ali Y等[32]合成了2-亚氨基-4-噻唑烷酮衍生物(化合物26,见图7),并评估了其体内外抗炎活性。结果表明,化合物26具有显著的COX-2抗炎活性和良好的选择性。乙酸诱导的痛觉实验表明,化合物26表现出与吲哚美辛相当的痛觉抑制作用,且不会引起胃溃疡和上皮组织损伤。

图7 化合物22~26的结构式Fig.7 Structures of compounds 22-26
2.6吡啶类 Renard J F等[33]用吡啶取代尼美舒利的硝基苯环合成了一种新型的化合物27(见图8),使用人全血模型在体外评估了该化合物对COX 2种同工型的抑制活性。结果显示,化合物27具有比尼美舒利和塞来昔布更有效的COX-2抑制作用和更高的选择性。Abdelgawad M A等[34]制备的嘧啶-吡啶杂化合物28(见图8)显示出较好的COX-2抑制作用。角叉菜胶诱导的大鼠足水肿实验显示,化合物28在给药2 h后显示出高于塞来昔布抑制水肿的作用。构效关系表明,苯环上供电子基团比吸电子基团取代的化合物显示出更好的水肿抑制作用。

图8 化合物27~28的结构式Fig.8 Structures of compounds 27-28
2.7咪唑类 Almansa C等[35]合成了1,5-二芳基咪唑类化合物29(见图9),体外抗炎实验表明,该化合物具有良好的COX-2抑制作用和选择性。通过大鼠气囊炎症模型实验评估该化合物的体内抗炎活性,结果表明,化合物29的炎症抑制率达到了98.2%。此外,该化合物在痛觉测试中表现出显著镇痛作用。Kiani A等[36]合成了5-取代的1-苄基-2-(甲基磺酰基)-1-H-咪唑(化合物30,见图9),并进行COX-2抑制作用评估。构效关系研究表明,取代基对化合物COX-2抑制作用的贡献排序为:-OCH3>-Br>-NO2>-H>-Cl。

图9 化合物29~30的结构式Fig.9 Structures of compounds 29-30
2.8生物碱类 Graziano A C E等[37]从红海藻中分离得到新型的偶氮酰基吗啉酮生物碱,与抗氧化剂丁基化羟基茴香醚、丁基化羟基甲苯和α-生育酚相比,化合物31(见图10)具有显著的1,1-二苯基-2-三硝基苯肼(DPPH)清除活性。体外抗炎实验表明,化合物31与NSAIDs相比显示出强效的COX-2和5-COX抑制作用以及与阿司匹林相当的选择性。角叉菜胶引起的大鼠足水肿实验表明,化合物31在给药6 h内能够保持明显的抑制水肿作用。白屈菜红碱(化合物32,见图10)是一种来源于白屈菜植物和其他罂粟科白屈菜属的季铵苯并[c]菲啶生物碱,具有广泛的药理作用。体外抗炎实验表明,化合物32具有较强的COX-2抑制作用。甲醛引起的大鼠足水肿实验表明,该化合物与对照组相比能够显著且剂量依赖性地抑制水肿,并且在给药3 h后达到最大抑制率。此外,在镇痛实验中,化合物32明显抑制乙酸诱导的大鼠扭体反应并减少其扭体次数[38]。Yang M H等[39]报道了从薯蓣(Dioscoreaopposita)根状茎的氯仿层分离的2个化合物,并评价了它们清除DPPH自由基、抗氧化的能力和体外COX-2抑制活性。结果表明,化合物33(见图10)能有效清除DPPH自由基,化合物34(见图10)显示了对超氧自由基的抗氧化活性,此外,化合物33和34均表现出显著的COX-2抑制作用。

图10 化合物31~34的结构式Fig.10 Structures of compounds 31-34
2.9香豆素类 Z Ju等[40]研究了3S-5,8-二羟基3-羟基甲基二氢异香豆素的体外抗炎活性,表明化合物35(见图11)具有显著COX-2抑制活性。Revankar H M等[41]报道了2个分别结合噻唑啉和噻唑烷酮部分的香豆素衍生物,化合物36和37(见图11)与参比药物塞来昔布相比,对COX-2表现出更高的亲和力和选择性。体内抗炎实验表明,在角叉菜胶诱导的大鼠足水肿测定中,化合物36和37的肿胀抑制率分别为96.73%、90.00%,并显示出较好的胃肠道安全性。

图11 化合物35~37的结构式Fig.11 Structures of compounds 35-37
2.10黄酮类 Bashir R等[42]报道的6-氯-7-甲基-3′,4′-二甲氧基黄酮(化合物38,见图12)与参比药物塞来昔布相比,具有更高的抗炎活性和优良的胃肠道安全性,此外,化合物38对COX-2的选择性几乎是塞来昔布的2倍。角叉菜胶诱导的大鼠足水肿实验评估了该化合物的体外抗炎活性,结果表明,以0.05 mmol·kg-1的剂量给药3 h后,水肿抑制率达到了100.00%,并且在给药的第5小时仍然保持较高的抗炎活性。Maicheen C等[43]评估了一系列作为潜在COX-2抑制剂的色酮衍生物,化合物39和40(见图12)与塞来昔布相比具有更高的体外抗炎活性。化合物39具有显著的COX-2选择性,其选择性与塞来昔布相似。分子对接研究表明,化合物40色酮部分的C-3位取代基位于疏水口袋中,相当于塞来昔布的对甲基苯基基团。而色酮母核位于侧面精氨酸的侧口袋中,与苯磺酰胺基团相似。说明色酮母核C-3上具有芳香取代基可能表现出与塞来昔布同等优秀的COX-2抑制作用。见图13。

图12 化合物38~40的结构式Fig.12 Structures of compounds 38-40

图13 化合物40与COX-2的结合图注:橙色:化合物40;绿色:塞来昔布;蛋白库编码(PDB ID):3LN1。Fig.13 Binding of compound 40 with COX-2Notes:Orange:compound 40;Green:celecoxib;protein library number (PDB ID):3LN1.
综上所述,炎症是人类防御机制中必不可少的部分,许多疾病的发展都与炎症相关。在过去的几十年中,NSAIDs被广泛用于治疗炎症相关疾病,但严重的胃肠道不良反应极大地限制了其临床应用。选择性COX-2抑制剂的发现为炎症相关疾病的治疗提供了新的选择。目前,COX-2已成为癌症、神经退行性疾病以及神经精神疾病的关键靶标。已有证据表明,选择性COX-2抑制剂塞来昔布在重度抑郁症和癌症的临床治疗中得到了有效应用,并且显示出良好的治疗效果。然而,目前只有几种选择性COX-2抑制剂应用于临床,大量具有COX-2抑制作用的化合物还处于动物实验研究中,寻找具有临床应用价值的潜在抗炎药依然是当今的研究热点。本文综述了几类化学合成及天然来源的COX-2抑制剂,将其运用到新药的设计中具有重要意义。
猜你喜欢
电气技术(2022年5期)2022-05-23
吉林大学学报(理学版)(2022年1期)2022-01-21
汽车工程师(2021年12期)2022-01-18
大连民族大学学报(2021年5期)2021-11-15
昆明医科大学学报(2021年1期)2021-02-07
新课程·下旬(2019年7期)2019-09-17
第一财经(2019年8期)2019-08-26
发明与创新·中学生(2017年11期)2017-12-07
作文·初中版(2017年6期)2017-06-16
中国民族民间医药·上半月(2016年12期)2017-01-11
