
细菌最小基因组研究进展
2021-02-25 13:32李金玉杨姗崔玉军王涛滕越
遗传 2021年2期
李金玉,杨姗,崔玉军,王涛,滕越
综 述
细菌最小基因组研究进展
李金玉1,2,3,杨姗2,3,崔玉军2,3,王涛1,滕越2,3
1. 天津大学生命科学学院,天津 300110 2. 军事科学院军事医学研究院微生物流行病研究所,北京 100071 3. 病原微生物生物安全国家重点实验室,北京 100071
具有最小基因组的细菌只包含维持自我生命复制所必需的基因,其作为一种潜在的工业生产平台具有诸多优势。由于高通量DNA测序和合成技术的发展,目前已经构建了多种缩减基因组的菌株。本文首先介绍了最小基因组的概念,其次总结了细菌必需基因的相关研究进展,然后梳理了人工缩减与合成微生物基因组的相关工作,最后探讨了在设计和组装基因组的过程中遇到的技术障碍和限制,以期为人工合成基因组的实验与应用提供理论参考。
细菌;最小基因组;必需基因;合成生物学;人工设计
人们很早就认识到支原体属()等物种的基因组比大多数细菌要小得多[1~3]。虽然最初认为这些小基因组生物代表了生物进化的原始祖先,但到20世纪80年代,基于16S核糖体RNA序列的系统发育研究证实:[4]以及其他具有小基因组的胞内细菌,如立克次氏体科()中的一些物种[5],皆来源于具有更大基因组的细菌。20世纪初,基因组测序结果证实了这些发现,并在不同的细菌类群中发现了独立的通过缩减基因组进化的案例,如柔膜菌纲中的[6]、α-变形菌纲中的普氏立克次体()[7]、γ-变形菌纲中的蚜虫内共生菌()[8]和螺旋体纲中的伯氏疏螺旋体()[9]。这些微生物的基因组保留了满足其自身在宿主中生存的关键基因,证明不同细菌类群在特定情况下具有丢失多种基因的“能力”,这表明控制基因组缩减的机制和动力可能存在于多数细菌中。
随着全基因组测序技术的发展,通过对几个胞内共生细菌的基因组测序发现存在更小的基因组,这突破了早期测序达到的约500 kb (即500个基因左右)的下限;其中一些基因组还包括其他极端特征,如极快的基因进化、密码子重排和核苷酸组成的极端偏向性等[10,11]。这些变化的主要驱动因素包括较小的种群规模以及无性繁殖背景下的选择、突变和遗传漂变[11~13]。这类生物不仅编码很少的蛋白质,而且其高度修饰的蛋白质很容易发生错误折叠,需要大量分子伴侣以保证蛋白质具有正常功能[10]。尽管与宿主相互作用的关键基因得以保留,但其基因组通过持续删除特定基因而不断被缩减。共生生物和宿主的共适应可能促进一些基因丢失,但与从线粒体和胞质体向细胞核的基因转移不同,细菌基因向宿主基因组的转移似乎与细菌基因组缩减无关。
20世纪初,伴随重组DNA技术的出现,合成生物学可以设计并构建基于合成遗传回路和通路的新型高潜力生物系统。在合成生物学的“设计–构建–测试–学习”循环中,系统生物学为其设计步骤提供了关于复杂生物过程的完整信息。因此,借助系统生物学和合成生物学,可最小化细菌基因组,使其仅包含细胞复制和产品生产所必需的基因,并以此设计和构建可预测、高效和精于生产的细胞(图1)。本文首先介绍了细菌最小基因组概念,其次总结了必需基因的研究进展,然后回顾了在构建人工缩减与合成基因组方面的研究工作,最后探讨了在设计和构建缩减基因组的过程中遇到的技术障碍和限制,以期为人工合成基因组的实验与应用提供理论参考。
1 最小基因组
1.1 最小基因组的概念
最小基因组被定义为在无外界压力(营养丰富和无应激)条件下足以维持生命活动的基因集。许多实验研究进一步将其定义为在丰富培养基中支持纯种培养的基因集。因为大多数生物在大自然的生态环境中需要额外的基因,所需的基因集会随着环境条件的变化而有所不同。最小基因组概念出现和发展的时间与细菌全基因组测序时代相同,都始于20世纪90年代中期[14]。生物的基因组大小从几十万个碱基对到一千多亿个碱基对不等。其中,布赫纳氏菌(spp.)是一种胞内共生菌,与大肠杆菌()具有共同祖先;与其祖先相比,基因组缩减达75%,仅为250 kb[15,16],是自然界中基因组大规模缩减的一个明显示例。最小基因组不仅存在于布赫纳氏菌,也存在许多其他共生细菌中。是一种昆虫共生细菌,具有最小的自我复制基因组(112 kb)[17]。此外,本课题组还聚焦于最近发现的为食液昆虫提供营养的共生细菌中的极端微小基因组,包括来自4个远缘细菌类群的5个独立的极端基因组缩减实例,其基因组大小都不到生殖支原体基因组的一半(甚至不到1/4) (表1)。这些例子显示出源于生态位适应的基因组进化趋势。共生生物不需要环境条件响应相关的基因,因为其宿主提供了稳定的营养供应,并保护其免受恶劣环境变化的影响,因此,在长期进化过程中,这些不必要的基因已经从它们的基因组中移除。
许多研究表明,物种种群规模较小、无性繁殖[18~20]以及细菌所固有的基因删除偏倚[19,21],是基因组缩减的主要原因。由于特定宿主的限制和不同宿主中的菌株之间缺乏重组,导致了高度遗传漂变,使有益但非必需的基因失活和缺失。这种菌群结构的另一个影响是最初发现于和中的微小基因组基因序列快速进化,及其对蛋白质二级结构稳定性的影响[22,23]。基因组减少的早期阶段以一些较新进化的共生生物为代表,其特征是移动元件的增加,假基因的形成,多个基因组重排和染色体片段的缺失(图2)[24~26]。然而,在更早期进化的共生体中,如,移动元件和大多数假基因已被移除。

图1 设计、构建和测试最小基因组的研究
以合成生物学为基础的最小基因组的构建,包括设计、构建、测试、学习、应用的循环周期。A:通过不同方法确定物种的必需基因集,包括比较基因组学、转座子测序、反义RNA、CRISPRi等。B:构建最小基因组的过程,主要遵循自上而下,自下而上两种原则。C:对构建的最小基因组进行系统生物学研究,以测序技术、组学技术等为手段进行测试、学习、应用。
通过对不同生物基因库的计算分析[14,27~32],以及纯种培养生物的基因诱变破坏实验[33~36],可对最小基因组的基因完整性进行预测。这种诱变实验只限于可纯种培养的生物,如.,它的基因组是迄今为止纯种培养生物中最小的。研究表明,通用基因集很小,且不包括许多已被实验确定为特定生物所必需的基因。因此,不同生物可能以完全不同的方式完成基本生命活动。例如,通过运输而不是从头合成获得所需的化合物,或者使用不相关的基因和不同的途径使特定tRNA装载正确的氨基酸[37]。某些通用或接近通用的基因缺失并不致死[32]。不同的研究预测了不同的最小基因集,虽然这些基因的分布较广泛,但都包括参与细胞基本功能的基因。有些新陈代谢相关基因分布较广泛,在自然环境下对特定生物至关重要,但并非必不可少。
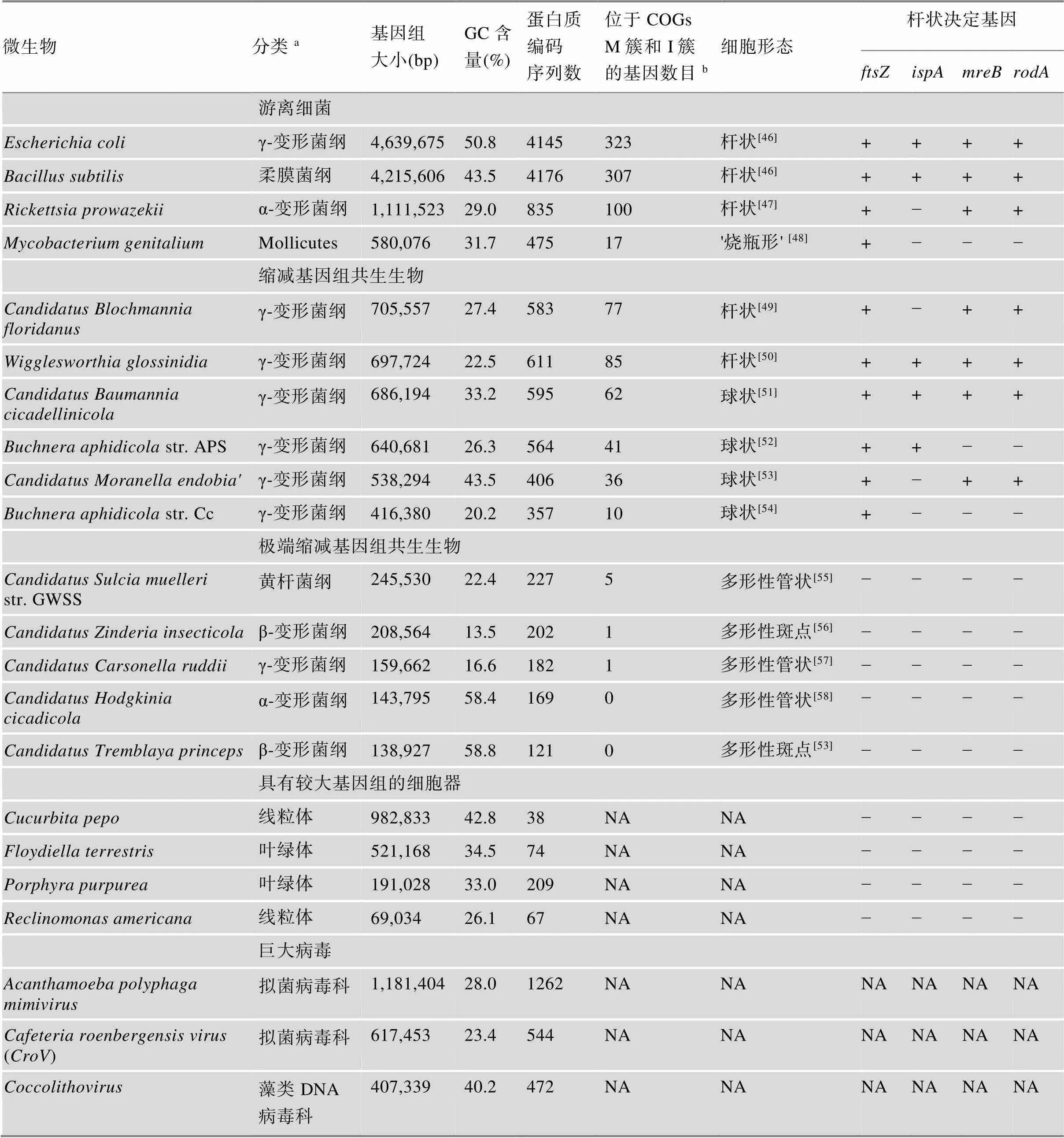
表1 具有代表性的细菌、共生生物、病毒和细胞器基因组的比较
a:指个体所属的纲,细胞器或病毒家族类型;b:蛋白质直系同源基因簇,即指位于蛋白质直系同源基因簇中M簇(细胞膜生物发生,外膜)和I簇(脂质代谢)的基因数;NA:不适用。
细菌基因组含有数百万个碱基对。例如,研究最广泛的模式生物大肠杆菌的基因组>5 Mb,包含4000多个基因,其中1000多个为未知功能的基因[38,39]。大肠杆菌可在多种环境下繁殖,如好氧和厌氧,以及不同营养物质、pH值和温度等。大肠杆菌的基因组中有许多基因负责处理环境压力和利用各种营养物质;然而,在环境确定的实验室条件下,不再需要某些应激反应基因;因此许多基因可以被删除,而不会对细胞生长产生负面影响[40]。同时,大肠杆菌基因组还编码了许多实验室培养和工业发酵不需要的基因,这些基因会导致能量和生物质前体的浪费,不必要的基因组片段的复制,以及功能冗余或无用的转录物、蛋白质和代谢物的合成。因此,删除这些不必要基因的生物体或可成为人工条件下产品生产的全新平台[41]。
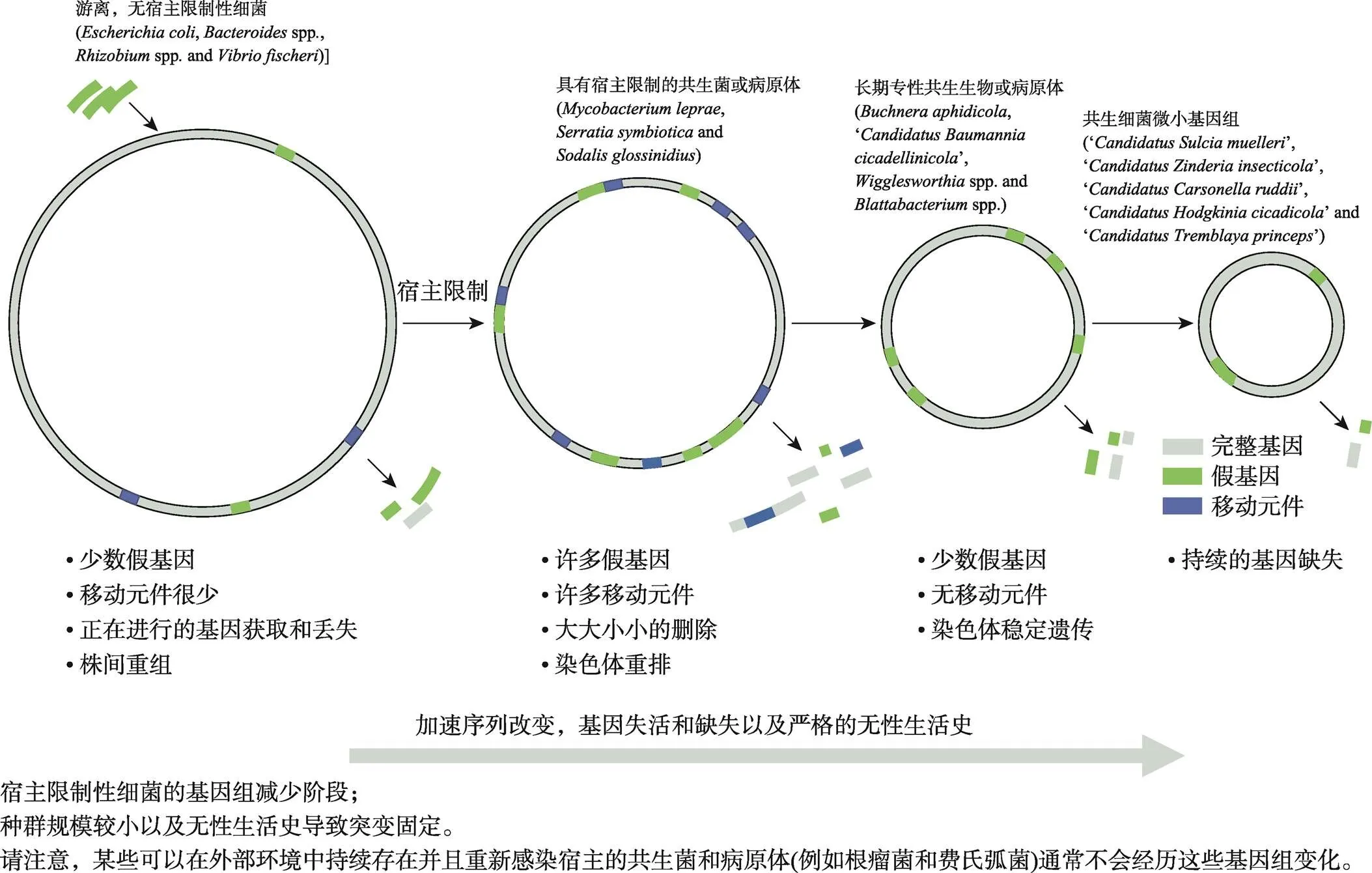
图2 细菌基因组的减少阶段及其进化特征
1.2 线粒体基因组简化
线粒体比较基因组学使人们了解线粒体祖先的基因组是什么样的,以及它包含的基因是什么。从它所包含的基因组的角度看,线粒体具有毫无疑问的细菌起源,起源于α-变形杆菌()[42]。因此,内共生假说认为真核细胞中的线粒体从细菌祖细胞通过共生进化而来。基于系统发育的分析表明,线粒体基因组进化的特征是某些谱系大量扩增,另一些谱系则极度减少和压缩,通过内共生基因转移将线粒体基因组中的许多初始遗传信息重新定位到细胞核[43]。α-变形杆菌基因组之间的比较表明,线粒体的细菌祖先包含约3000~5000个基因,线粒体基因组中直系同源基因的祖先簇的上限约为1700个,这些估计表明,从细菌共生体到原始细胞器的过渡过程中丢失了大约3000个基因[44]。可以预见,基因组学和蛋白质组学数据的不断增长将提供关于线粒体结构和功能的新的数据和见识,并将不断重塑和完善目前的线粒体进化理论。
1.3 构建和设计最小基因组
随着基因组学革命以及系统生物学的不断进步,合成生物学在过去10年中飞速发展,尤其是在基于系统和合成生物学的微生物底盘工程和改造领域。在系统生物学中,理想的底盘可代指具有简化的基因组且可以实现全部功能的生物,以及能够更有效地合成所需产物的代谢网络;在合成生物学中,底盘是指通过提供允许其运转的资源来容纳和支持遗传成分的生物。
构建合成微生物底盘包括两种方法:自上向下和自下向上。自上而下的方法是通过去除不必要的细胞基因来了解基因组架构并改善其特性的减少基因组大小的策略。基于大规模DNA分析的出现,通过对来自不同生物体的基因组进行比较分析,通常可以揭示对于细胞生命和相似和/或截然不同的代谢途径必不可少的基因。接下来,再通过不同的实验方法来实现缺失。工程设计和修改合成微生物底盘揭示生命的基本原理,是加强其在卫生医药和食品工业中的应用的最佳方法之一。有关这个主题的研究计划已在多个国家获得资助。“最小基因组工厂”(minimum genome factory, MGF)项目于2001年在日本启动,其目标是构建具有较小基因组的微生物以用于工业用途。基于比较大肠杆菌和布赫纳氏菌获得的非必需基因信息,Hiroshi等[45]通过删除大肠杆菌W3110中的大肠杆菌特异性基因构建了大肠杆菌MGF-01,其构建方法与其他缺失方法一致,即连续缺失结合P1转导。简言之,用含有和基因的DNA组件同源替换删除靶DNA序列。然后,用另一个DNA组件替换选择标记,用蔗糖进行负筛选获得无标记克隆。最后,在28个P1转导循环中积累了53个缺失,从而使总缺失长度达1.03 Mb。MGF-01的最终生物量是野生型亲本的1.5倍。据称,MGF-01相比亲本菌株葡萄糖利用效率更高,这是因为显著减少了过量的醋酸盐积累(从大肠杆菌W3110中的1.37 g/L降至MGF-01中的0.50 g/L)。此外,与野生型相比,MGF-01的苏氨酸浓度和产量分别提高了2.44倍和1.69倍,醋酸盐副产物则显著降低。MGF-01的这些意想不到的有益特性与其他具有与野生型祖先相似表型的缩减基因组大肠杆菌不同,但需要进一步研究MGF-01的差异性。
2 细菌必需基因
构建最小基因组,首先要阐明维持生命所必需的基因。20世纪90年代,只测得少数微小细菌的全基因组序列。Mushegian和Koonin等[59]对生殖支原体和流感嗜血杆菌()进行比较,表明这两种细菌都有相对较小的基因组,但表现出完全不同的进化轨迹。根据比较基因组学原则,在多种生物中保守的基因很可能有必需的功能。在生殖支原体和流感嗜血杆菌中,共发现240个直系同源基因;但此分析遗漏了几个编码细胞必需功能的基因,如磷酸甘油酸变位酶和核苷二磷酸激酶基因。这两个物种有不同且不相关的磷酸甘油酸变位酶,因此非同源基因偶尔可以取代古老的基因并破坏其保守性。最终,研究结果证明包括非直系同源基因取代在内,预测共有262个基因构成了两个物种的核心生物功能。尽管这两种细菌和它们的共同祖先之间经历了15亿年的进化,仍有约50%的基因被保存下来。
自2000年以来,已获得数万基因组序列。科学家比较了数百个基因组,以确定物种间普遍存在的基因,然而这样的基因并不常见。Brown等[61]比较了45个基因组,发现只有23个保守基因[60];Koonin等[62]报道了普遍存在于100个基因组的63个基因;Charlebois等[63]比较了14个门中的147个原核生物基因组,仅发现34个通用基因。虽然报告的保守基因数目不同,但都这些研究同时证明只有少数保守基因绝对不足以维持生命。因此,比较基因组学对必需基因的理论预测仅限于功能未知、非同源取代和具有数十亿年进化历史的基因。
除了计算预测,研究者还使用实验方法来探究必需基因。最简单的方法是从基因组中删除一个特定的基因位点,观察其致死效应。在迄今为止已知的任何自由生活的生物体中,生殖支原体具有最小的基因组(580 kb,编码517个基因),其中许多基因可能通过转座子作用而失活[64]。在枯草芽孢杆菌()中,通过对其基因组的79个区域进行随机突变[65],发现只有6个位点是必不可少的,大小约318~526 kb,与生殖支原体基因组大小相似。因此,人们设计了各种方法来确定细菌中的必需基因。如通过重组、转座子插入和反义RNA (asRNA)直接使单个基因失活。通过插入非复制质粒失活枯草芽孢杆菌中的单个基因,Kobayashi等[66]发现4101个基因中只有271个是必需基因。在大肠杆菌中,4288个开放阅读框(open reading frame, ORF)中的3985个可被敲除,表明剩余的303个ORFs对生命至关重要[67]。尽管靶向基因敲除研究提供了基因必需性的直接证据,但该方法耗时长,需要进行数千次删除实验。
为了克服这些限制,可采用基于转座子诱变失活的高通量方法。转座子是一种基因元件,可在基因组内随机移动并插入,破坏基因的正常功能。如果基因组中的必需基因被插入转座子,则此突变体不能存活。利用此特征,可以通过鉴定存活突变体中的转座子插入位点来区分必需和非必需基因。Glass等[68]和Hutchison等[69]分别利用1300和3000个突变体,在生殖支原体中构建了全基因组转座子插入图谱,发现265~382个必需编码序列(coding sequences, CDSs)。由3000个唯一插入位点组成的转座子插入图谱,其平均分辨率约为每200 bp一个插入位点。此分辨率无法检测功能性RNAs (如tRNAs和ncRNAs)等小遗传元件的必需性。此外,一些必需基因在3ʹ端对转座子插入检测具有抗性,因为短截尾或延伸并不影响其功能。为了规避这一限制,研究者发明了一种通过asRNAs使基因失活的方法[70]。与转座子不可逆的基因破坏不同,利用asRNAs可以特异性敲低某个基因。因此,一旦构建出asRNA文库,研究者可以在多种环境条件和迭代过程中评估基因必需性或适合性,而无需重复构建敲除菌株或转座子突变体。
转座子插入位点的鉴定依赖于单个克隆的分离和现有的高通量测序技术的发展;通过与高通量测序技术相结合,可以并行识别多个插入位点。因此,转座子诱变与下一代测序技术(Tn-Seq)相结合,即利用转座子诱变以更高的分辨率检测必需基因。对2×105大肠杆菌转座子突变体文库的统计分析,共鉴定出620个必需基因[71]。Tn-Seq和单个基因敲除研究之间的差异可能源于细胞增殖。一个重要基因,即使不是严格意义上的必需基因,其失活也会导致严重的生长缺陷;在细胞繁殖过程中,这种缺陷可能未被充分表达或逐渐消失。此外,不同的统计临界值和实验条件也可能导致这种差异。
最后,基于成簇规律间隔短回文重复序列(clustered regularly interspaced short palindromic repeats, CRISPR)引领的技术革命,催化失活Cas9 (dead Cas9, dCas9)可以在转录水平抑制靶基因的表达[72]。由于CRISPR系统的特异性只取决于嵌合单导向RNA (sgRNA)中20 nt的原间隔序列,因此易于通过DNA合成构建大规模全基因组sgRNA文库。利用全基因组CRISPR干扰(CRISPR interference, CRISPRi),通过约59 000个sgRNAs抑制所有基因,在大肠杆菌中鉴定出379个必需基因[73]。利用CRISPRi技术估计的必需基因数量略大于其他方法估计的数量。在细菌中,许多基因是多顺反子转录的,故前导多顺反子的破坏会使操纵子中包含的下游基因失活。因此,即使前导基因是非必需的,其转录抑制也会导致下游必需基因的致死效应,即多顺反子结构导致必需基因的高估。利用目前的高通量DNA合成技术,可以高效合成庞大的sgRNA文库,并将其用于鉴定具有更大基因组的生物(如人类)的必需基因[74,75]。
总之,人们已经采用多种方法来阐明各种生物体中的必需基因。尽管不同方法得到的必需基因的确切数目不同,但500个基因被认为足以维持生命。利用单基因敲除研究、Tn-Seq和CRISPRi直接检查单个基因是否必需无法规避各种技术固有的局限性。因为这些方法依赖于单个基因的移除或失活,而同时失活两个以上的基因从未被测试过。例如,假设两个基因在大肠杆菌中具有相同的基本功能,这两个基因可以单独删除,因为另一个基因可以恢复其功能;但是,两个基因同时失活则产生致死效应。即使在最简单的细菌生殖支原体中,构建双基因敲除库也需要25万个菌株。考虑到构建约4 000个大肠杆菌单基因敲除菌株所需的巨大时间和费用成本[40],需要采用新的技术来探索基因高阶组合的必需性。
3 人工缩减基因组微生物
大肠杆菌是研究最广泛的模式生物,已为其构建了多种缩减基因组,缺失大小从300 kb到1.38 Mb不等(表2)。大肠杆菌K-12菌株的基因组约为4.64 Mb,缺失大小约为原始基因组的6.8%~29.7%。下面简介了一些目前构建的缩减基因组大肠杆菌,还回顾了其他物种中基因组缩减案例。
3.1 大肠杆菌CDΔ3456
2002年有两例关于缩减基因组大肠杆菌的报道。其中之一是Yu等[76]报道的CDΔ3456,缺失片段大小超过300 kb。该菌株构建过程中,位于位点特异性重组酶识别的两个位点之间的一个较大的基因组区域被删除。使用转座子将位点预先插入到基因组中的任意位置。具体方法是,用两种抗生素抗性基因盒构建两个转座子突变库,并定位所有转座子插入位点。筛选转座子分别定位在目标缺失区域一端的两个突变体,其中包含两个不同的标记基因,并通过P1转导将其基因组融合。使用两种不同的抗生素抗性基因标记来筛选成功转导的细胞。然后,融合基因组中的两个位点通过Cre重组酶重组,产生较大的靶向缺失。由此构建了6个缺失菌株,并进一步利用P1转导将多个缺失累积到一个基因组中。在此过程中,某些基因组区域可以单独从基因组中删除,但是特定的基因组合不能同时删除,这种关系被称为合成致死,其原因可能是存在基因重复或直系同源物[77]。当两个基因的一个拷贝被删除时,另一个拷贝可以维持生物体正常功能,而双突变体则不能。为了避免合成致死组合,4个区域被整合在CDΔ3456中,该菌株缺少287个ORFs,其中包含179个未知基因,以及与组氨酸生物合成、菌毛和数个转运蛋白相关的基因,最终获得克隆的生长速率与亲本大肠杆菌相当。
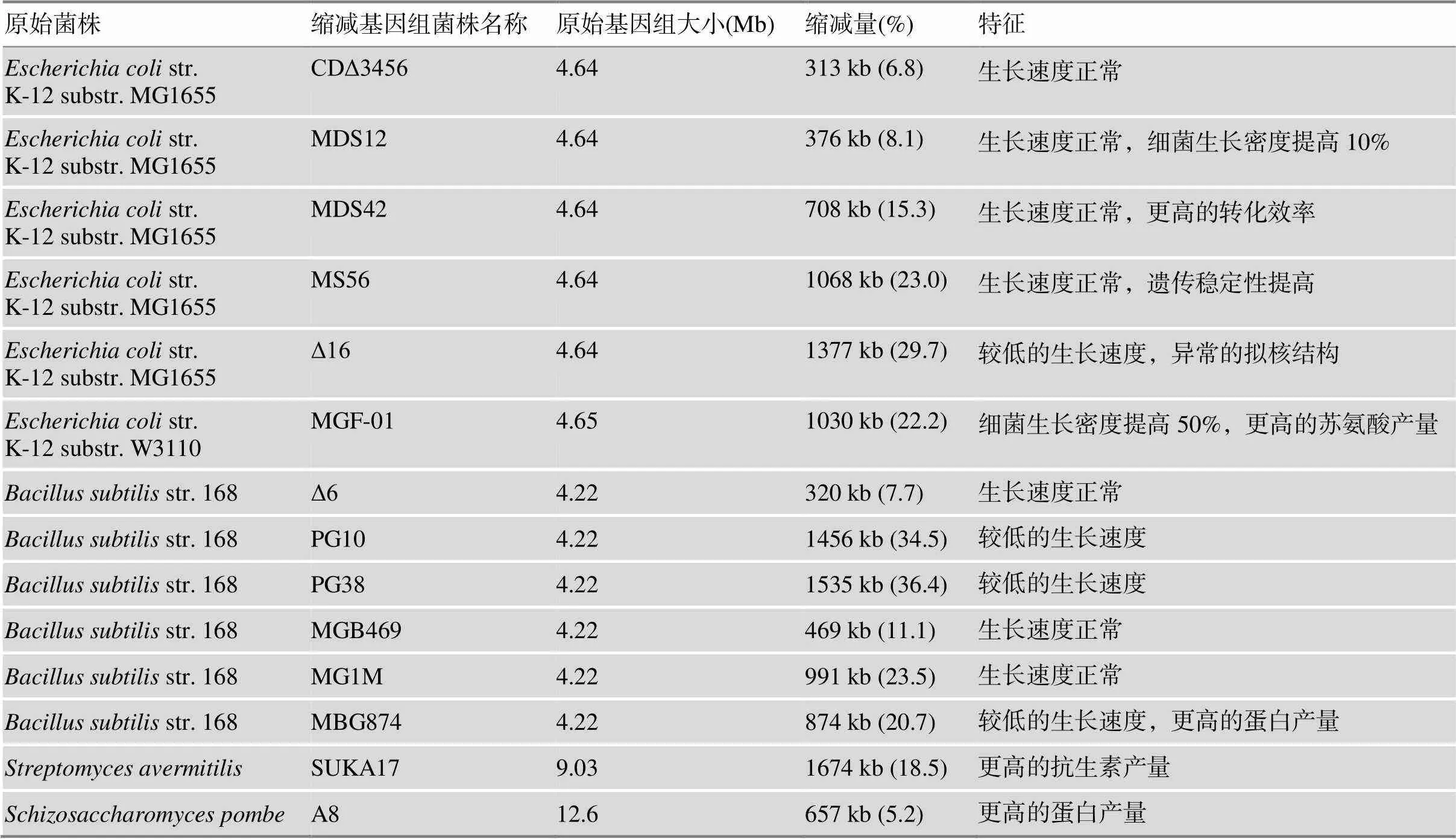
表2 缩减基因组大肠杆菌及其特征
3.2 大肠杆菌MS56及其近缘菌株
2002年报道了缩减基因组大肠杆菌MDS12,其缺少大肠杆菌K-12菌株的12个K-岛[78]。K-岛是K-12通过水平基因转移获得的基因组区域。K-岛含有非必需基因,如原噬菌体和转座子。利用I-I巨核酶和双链断裂修复系统,用无痕删除法将12个K-岛依次删除。具体方法是,将含有氯霉素抗性基因的DNA组件引入大肠杆菌MG1655,以取代同源靶区。虽然靶标被删除,但下一轮删除需要去除抗性基因,因此采用I-I将该基因删除。
之后,双链断裂被RecA修复,形成无痕缺失株。经过12次迭代删除和P1转导,删除长度合计为376 kb,包含409个ORFs。由于被删除的基因没有必需功能,MDS12的生长速度和DNA转化效率与原始菌株无差异。MDS12的最终细胞密度比野生型大肠杆菌高约10%。在MDS12中,节省的能量和物质可以转化为生物量,显示出缩减基因组的优势。
4年后,Pósfai等[79]报告了大肠杆菌MDS41、42和43,它们是MDS12的后代,含有额外的缺失(与野生型相比,总缺失为663~708 kb)。MDS42不含任何可转位和插入序列(insertion sequence, IS)元件。据报道,在大肠杆菌中,20%~25%的突变与IS元件有关[79]。MDS42未显示IS介导的基因失活。利用该特性,MDS42可稳定复制携带毒性ORFs的质粒DNA,且基因组构成稳定。大肠杆菌有一个沉默的操作子,能够利用水杨苷;因此,通常条件下,大肠杆菌不利用水杨苷作为其唯一碳源。当使用水杨苷作为唯一碳源培养时,MDS41的操纵子激活率低于MG1655 (8%)。此外,MDS42产生的苏氨酸比野生型大肠杆菌多80%以上[80]。
成功构建MDS菌株之后,Park等[81]在MDS42中又引入了14个缺失,以此构建大肠杆菌MS56。除MDS42的缺失外,还删除了非必需基因和遗传元件,如氢化酶、菌毛样黏附素和厌氧呼吸酶,基因组缩减总量达1.07 Mb。编码抑制大肠杆菌生长的人蛋白异源基因可在MS56中成功表达,不会有任何IS介导的失活,而正常含IS的大肠杆菌中会迅速发生失活。
3.3 大肠杆菌Δ16
Hashimoto等[82]构建了大肠杆菌Δ16,其基因组缩减量为1.377 Mb (占其原始基因组的29.7%),是报道的最大缩减量。与其他缩减基因组一样,Δ16的构建方法也是同源重组连续缺失结合P1转导。Hashimoto等[82]采用正筛选和负筛选两种方法进行基因删除。首先,构建靶区序列末端同源DNA组件。该盒还含有三个基因:氯霉素乙酰转移酶()、和。将此DNA组件导入后,用氯霉素选择目标基因组区域被此组件取代的克隆。然后,用新的同源DNA组件取代该序列,接着用和基因进行负筛选,使细胞对链霉素和蔗糖敏感。通过P1转导将16个无痕缺失进行组合,获得最终菌株Δ16,累计缺失1.377 Mb。在连续缺失过程中,倍增时间从MG1655的26.2 min依次增加到Δ16的45.4 min,细胞形状变长变宽,拟核呈不规则分布,其复制与细胞分裂不同步。
3.4 枯草芽孢杆菌PG38及其近缘菌株
枯草芽孢杆菌是研究最广泛的革兰氏阳性菌之一,因其具有蛋白分泌系统而成为各种蛋白的优良生产宿主。Westers等[83]通过去除6个基因组位点,包括聚酮、蛋白质抗生素生物合成、原噬菌体和原噬菌体样元件相关基因,构建了基因组缩减枯草芽孢杆菌Δ6。这些位点共包含332个基因(320 kb)。通过使用整合质粒pG + host4整合并切除选择标记实现基因删除[84]。基因组缩减枯草芽孢杆菌Δ6在细胞生理上没有明显变化。与亲本菌株相比,该菌株的生长速度、葡萄糖/醋酸盐代谢通量、异源蛋白分泌和生物量均相同。出乎意料的是,尽管没有删除与细胞运动相关的基因,Δ6在琼脂糖平板上显示出细胞运动性增加。基因组缩减菌株偶尔会出现此类意外表型,说明对即使是微小细菌的基因组理解也是有限的[85]。
枯草芽孢杆菌Δ6基因组被进一步缩减,构建形成PG10和PG38。Reu等[86]构建了两个独立的基因组缩减菌株PG10和PG38,分别包含88和94个迭代缺失,缺失部分包含产孢、运动、抗生素合成和次生代谢相关的非必需基因。在连续缺失过程中,中间菌株逐渐丧失DNA整合所需的感受态。因此,在基因组中引入了额外的感受态蛋白(ComK和ComS),以有效导入缺失所需的DNA。两种衍生菌株的生长速度较慢(倍增时间延长约40%),细胞形态呈长丝状。虽然这两个菌株的生长速度都有所降低,与之前认为细胞存活所需的基因组相比,它们的基因组缩减比例是迄今为止最大的(1.46和1.54 Mb;超过了它们原始基因组的1/3)。
3.5 枯草芽孢杆菌MGB874及其近缘菌株
Ara等[87]基于枯草芽孢杆菌168构建了一株缩减基因组枯草芽孢杆菌MGB469。缩减菌株缺少9个与原噬菌体和原噬菌体样元件相关的基因组位点和2个抗生素合成基因(巴斯他汀和聚酮)。首先从枯草芽孢杆菌的基因组中逐一删除这些位点,以确认其不包含必需基因。然后,依次合并删除,使删除区域达469 kb,由此得到菌株MGB469,其生长速度和蛋白质产率与原始菌株无差别,从而达到进一步缩减基因组的目的。该文作者发现一个可以增加蛋白质产率的基因组缩减位点,最终构建出比原始菌株具有更高生产率的基因组缩减菌株。从MGB469中进一步删除单独缺失时增加纤维素酶产率的6个基因组位点,构建了总基因组缩减991 kb的菌株MG1M。然而,MG1M中纤维素酶和蛋白酶的生产没有发生明显的变化。
由于枯草芽孢杆菌MGB469和MG1M的表型与野生型相当,并且在进一步基因组缩减后没有产生有益表型,Morimoto等[88]重新利用中间菌株MGB469构建具有优势特征的新型基因组缩减枯草芽孢杆菌。该文作者测试了包括原噬菌体和次级代谢基因在内的74个基因组区域的缺失。在63个可发生缺失的区域中,11个连续缺失被引入MGB469,从而得到缺失长度为874 kb的枯草芽孢杆菌MGB874。虽然MGB874具有正常的细胞形态和拟核结构,但其生长速度降低至野生型枯草芽孢杆菌的70%。与枯草芽孢杆菌的其他缩减基因组不同,MGB874产生的纤维素酶和蛋白酶比枯草芽孢杆菌168分别提高1.7倍和2.5倍。转录组研究发现了许多转录组水平的基因表达变化,如产孢、降解酶分泌和σ因子,推测是其生产率提高的原因。MGB874证明具有缩减基因组的菌株可作为工业蛋白生产的优良宿主。
3.6 阿维链霉菌(Streptomyces avermitilis) SUKA17
放线菌门中的链霉菌属是一类工业和临床应用中重要的放线菌,已知其可产生抗生素等大多数具有生物活性的次级代谢产物。链霉菌属基因组在细菌中相对较大,其染色体DNA呈线性而非环状。基于应用最广的工业菌种之一阿维链霉菌,Komatsu等[89,90]构建了菌株SUKA17。阿维链霉菌中含有20多个次级代谢物生物合成基因簇,主要位于基因组的末端,称为亚端粒区。对灰色链霉菌()、天蓝色链霉菌()和阿维链霉菌进行比较基因组学研究,揭示了位于基因组中心的必需核心基因。使用同源重组和Cre介导的重组,从阿维链霉菌中删除亚端粒区主要次级代谢相关基因(包括萜烯代谢)。与大肠杆菌和枯草芽孢杆菌不同,阿维链霉菌不易发生发重组。因此,两个位点首先通过同源重组从环状DNA模板导入到基因组中。然后,通过Cre介导重组删除基因组中大片段目标位点。该缺失包括1272个ORFs的基因组区域,总长度为1.67 Mb。由于其基因组较简单,无需承担不必要的次生代谢物合成,含有异源基因簇的SUKA17比原生灰色链球菌和棒状链霉菌()产生更多的链霉素和头霉素C。
4 合成基因组学
4.1 基于合成基因组学的微生物设计与改造
上面讨论的缩减基因组都是使用自上而下的方式对基因组进行缩减而构建的。现今,研究者已实现从头合成病毒、噬菌体和细菌基因组。借助基因组编辑和合成工具,可以进行全基因组水平上进行工程设计。研究者已实现设计并构建基于合成遗传回路和通路的新型高潜力生物系统。借助系统生物学和合成生物学而发展的合成基因组学,可最小化细菌基因组,从头开始合成基因组可以对基因组结构和功能进行前所未有的修饰,获得对生命基本原理的新见解并致力于设计有价值的应用,例如设计并构建可预测的、高效的或是精于生产的细胞(图3)。
自1995年关于生殖支原体基因组大小为525个基因的第一份报道以来,该细菌和该属的其他成员已成为探索赋予生命最少数量的细胞成分的最有用的试验台。事实上,到目前为止,某些支原体菌株已成为我们最了解的生物系统,其所有组成部分及其相互作用的各个方面都经过了非常详细的探索和量化。里程碑式的工作包括通过转座子诱变检查每个基因的必要性,实现支原体基因组的完全化学合成等。
支原体是小基因组细菌,其将成为探索生命所需的最低限度的基因组大小的理想平台。Gibson等[91]构建的支原体JCVI-syn1.0含有一个完全化学合成的基因组,即略加修饰的1.08 Mb蕈状支原体()基因组的复制体。通过转座子诱变阐明蕈状支原体的必需基因后,合成了最小基因组蕈状支原体JCVI-syn3.0[92](包含473个基因,总长度为531 kb)。进一步的实验验证表明,基于分子生物学的系统知识以及有限的转座子诱变数据相结合的初始设计未能产生活细胞。随后,改良的转座子诱变方法揭示了稳健生长所需的一类准必需基因,这解释了其最初设计的失败。最后,研究者通过设计,合成和测试的3个循环(保留了基本必需的基因)产生了JCVI-syn3.0,相对于其亲本菌株而言,基因组的净基因组大小减少了约50% (从约1079 kb减少到531 kb),并将基本生物学信息精简到了473个基因,其基因组比自然界中任何自主复制的细胞都要小。虽然JCVI-syn3.0的倍增时间是JCVI-syn1.0的3倍(~180 min),但远小于具有天然最小基因组的生殖支原体(~16 h)。尽管全基因组合成需要漫长的时间和巨大的费用[93],但无疑是基因组缩减的一种极有吸引力的替代方法。
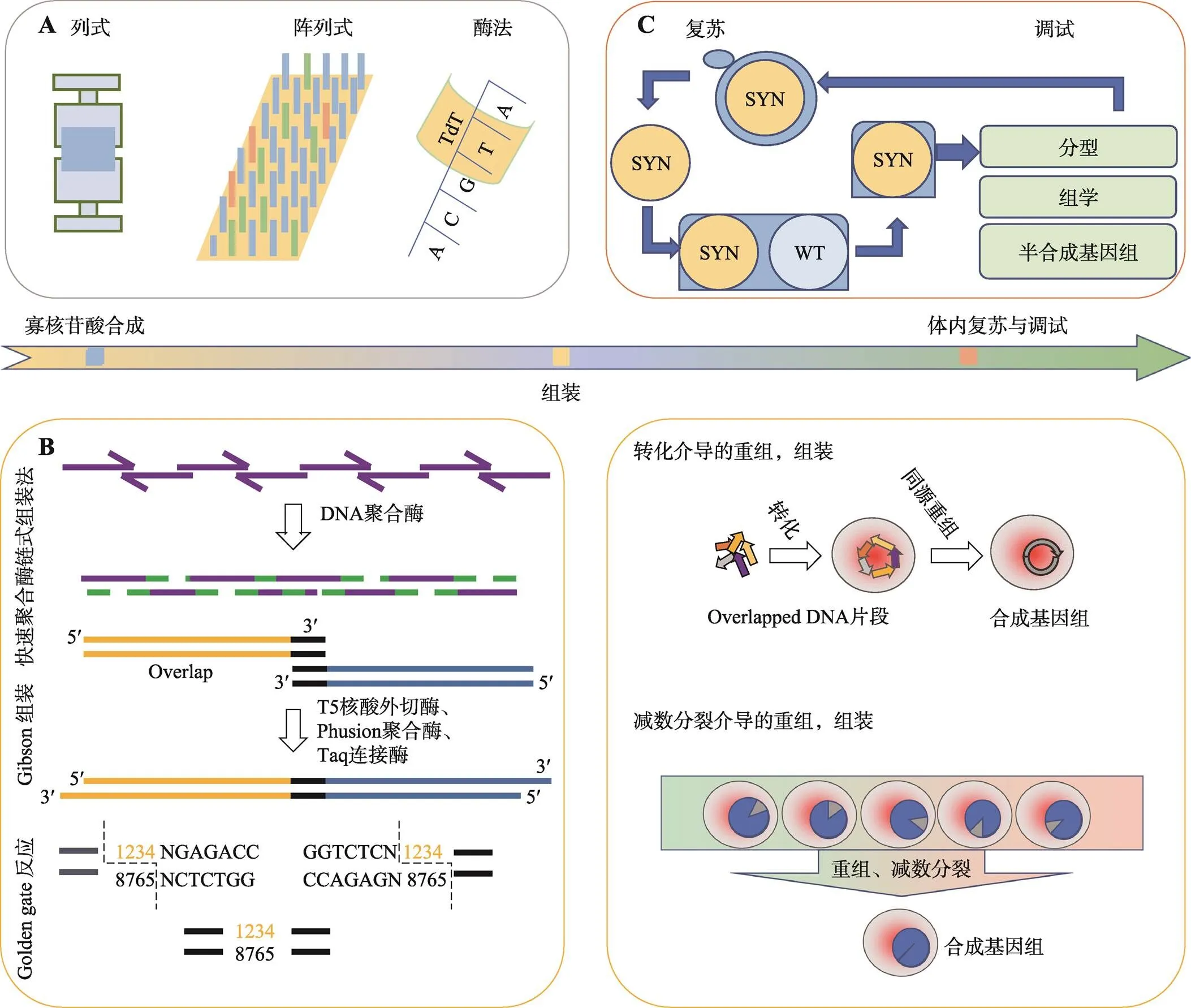
图3 合成基因组的过程:从设计序列到体外组装到体内复苏
A:在体外进行寡核苷酸的合成。常用方法包括列式合成、阵列式合成、酶法合成等。B:通过PCA、Gibson组装等进行体外组装,或是导入细胞内进行由转化以及减数分裂介导的重组、组装。C:最后进行分型、调试。
JCVI-syn3.0是一个多功能平台,可用于研究生活的核心功能并探索全基因组设计。最小细胞的概念乍看之下似乎很简单,但基于实验的实践验证却变得更加复杂。除了必需和非必需基因外,还有许多准必需基因,它们对生存力不是绝对关键,但对于强劲的生长却是必需的。因此,在最小化基因组的过程中,需要在基因组大小和生长速率之间进行权衡。JCVI-syn3.0近似为最小细胞基因组,为最小基因组与实验生物可行生长速率间的折中,它保留了几乎所有与大分子合成和加工有关的基因。出乎意料的是,它还包含149个生物学功能未知的基因,表明存在着生命必需的未发现功能。
4.2 大肠杆菌基因组密码子的简化
大自然使用64个密码子编码来自基因组的蛋白质合成,并从多达6个同义词中选择1个有义密码子编码每个氨基酸。同义密码子选择具有多种重要作用,且许多同义替换是有害的。合成基因组不仅模仿模板DNA,而且允许遗传密码重编程。 Lajoie等[94]将80个大肠杆菌菌株中42个高度表达的必需基因中13个稀有密码子的所有实例与编码相同氨基酸的密码子交换,显示了在活细胞中以全基因组规模进行编码的可行性。
生命科学的技术进步加快了我们处理基因组编码信息的能力,包括接合组装基因组改造(conjugative assembly genome engineering, CAGE)技术等。通过定义的同义密码子对目标密码子进行全基因组取代,可以减少用于编码规范氨基酸的密码子数量,最后,通过重新编码18 214个密码子,创建了一个具有61个密码子,基因组大小为4 Mb的大肠杆菌变体,其利用59个密码子编码20个氨基酸,并能够删除以前必不可少的转移RNA[95]。使用减少数量的有义密码子(59)编码20个必需氨基酸产生的生物体表明,生命可以在减少数量的同义有义密码子下运转。研究者开发的将设计的基因组分为多个片段、部分,通过Rexer、定向缀合等方法进行融合,最后进行无缝和强大的集成来实现设计的策略,为将来的基因组合成提供了一个蓝图。
4.3 DNA组装技术
作为合成生物学的基石,DNA组装过程允许使用被定义的、标准化的、特征明确的组件构建新颖的生物学系统。实际上是将多个DNA片段首尾相连的一种物理方式,从而创建目标高阶装配,然后将其连接到载体。采用限制酶切消化和逐个元素克隆的传统技术既耗时又低廉成本。因此,人们正在付出巨大的努力来开发更好的克隆策略和DNA组装技术,以使多基因系统能够被更快,更有效地构建。这样,构建具有复杂遗传功能的菌株将变得更加容易。这些新技术还有助于提高重组菌株的活力、可转化性或是稳定性。
DNA组装技术的加速使用使的得研究人员能够进行更复杂的合成项目,常见的组装方法除了上面提到的Gibson 组装,还有Golden Gate组装(GG)。Golden Gate模块化克隆系统利用II型限制酶建立了标准化且可互换的DNA部分文库,随后将其一步一步组装到一个预先设计的4nt悬臂支架。目前,Golden Gate组装已成为许多基因组编辑试剂盒的核心,许多实验室已经具备在单个反应中按定义的顺序装配许多不同零件的能力。GG的优点之一是它允许组合组装,可用于高效地生成库。此外,使用GG的启动子改组策略用于筛选表达的每个转录单位的最佳启动子–基因对,然后使用最佳启动子组合工程改造脂质过量生产菌株。
5 缩减基因组的障碍与挑战
解析突变和选择对细菌GC含量的不同影响一直是个难题。1962年,研究者提出一个模型,将基因组中GC平均含量描述为由(G或C)→(A或T)和(A或T)→(G或C)碱基替换率差异所驱动的严格中性突变的过程函数。之后,人们将细菌类群之间基因组GC含量的变异归因于谱系特异性突变模式和对各种全基因组特性的选择[96]。最近发表的两篇论文指出存在一种固有且普遍的(G或C)→(A或T)突变偏倚,并表明有利于高GC含量的选择过程是决定细菌基因组碱基组成的主要因素[97,98]。
缩减的细菌基因组往往具有更高的AT含量,且有时AT含量会急剧增加。具有最高AT偏倚的细胞基因组来自共生菌‘’ (β-变形菌,基因组为209 kb,GC含量为13.5%[99])和‘.’ (γ-变形菌,GC含量为16.6%[100])。已提出几种假设来解释严格内共生菌的AT偏倚,包括选择或群体遗传结合突变模式变化[101~103]。微小基因组倾向于删除许多参与DNA修复的基因,这可能导致更多的A或T突变,因为脱氧核糖核酸损伤,如胞嘧啶脱氨和鸟嘌呤氧化,往往会导致(G或C)→(A或T)[104]。穆勒棘轮效应(Muller’s ratchet)和纯化选择压力减轻将产生更多轻微有害突变,此类突变偏向A或T突变,并在种群中得到固定。这几种因素的共同作用使基因组GC平均含量移向AT含量较高的新平衡点。
值得注意的是,两个最小的细菌基因组,.[105](基因组GC含量为58.8%,四重简并密码子第三位的GC含量为66.6%)和.[105](α-变形菌,基因组大小为144 kb,基因组GC含量为58.4%,四重简并密码子第三位的GC含量为62.5%),打破了极端缩减基因组和低GC含量之间原本普遍的联系。在.中,缩减基因组表现为偏向GC的突变压力,因为在蛋白质序列水平上,四重简并密码子第三位几乎没有选择压力[106]。由此提出一种假设,即在基因组缩减的过程中,.以某种方式维持了自由生活α-变形菌中的高GC含量突变偏倚。通过后续研究.和.的突变方向,可以确定它们的GC突变偏倚是否具有普遍性,或它们是否像大多数(或所有)其他细菌一样,存在AT突变偏倚[97,98]。因此,其独特基因组组成的一个最明显的解释是全基因组范围内偏向GC的选择压力。
在细胞和细胞器基因组中,多种遗传密码独立于“通用”密码[107],UGA从终止密码子重新分配到色氨酸密码子是最常见的密码变化之一。在细菌中,这种密码子改变发现于柔膜菌纲的一个谱系中[108],这也是直到最近唯一报道的细菌编码重组事件。对来自昆虫共生细菌的微小基因组进行测序,发现了这种终止子–色氨酸重编码的两个新案例:.[107]和.[99]。这是柔膜菌纲谱系以外的细菌中唯一发现的密码子重新分配的两个案例,而.是唯一已知的在高GC含量的基因组中发生UGA密码子重新分配的案例。这一发现,以及基于几种线粒体基因组对密码子重新分配机制的分析[109],似乎与广泛引用的观点不符,即低GC含量是密码子重新分配的先决条件(“密码子捕获”假说)[110~112]。McCutcheon等[106]设想了一种基于“模糊翻译”[109]的机制,其中tRNATrp突变允许色氨酸(UGG)和终止(UGA)密码子的混杂解码,从而允许通过持续的基因组缩减移除识别UGA密码子的肽链释放因子2 (RF-2;由编码)。在此假设中,UGA密码子重新分配是对关键基因缺失的一种遗传共适应,而不是为了使基因组(或翻译)更加精简的适应性事件[113],也不是通过GC含量偏倚导致的密码子丢失和重新分配的完全中性事件。
大部分缩减基因组菌株的生长速度与其原始菌株相当。其中一些菌株的生长速率和最终生物量甚至高于其亲本菌株。此外,缩减基因组还具有诸如更高的转化效率[79]和生物化学法生产[85]等有利特性。少数情况下,尽管与生长、细胞周期和形态相关的基因未发生改变,但缩减基因组表现出意想不到的表型,如生长迟缓和异常细胞形态。最近一项研究提出了一种实验室适应进化(ALE)技术,以改善缩减基因组大肠杆菌的生长表型[80]。对进化菌株的多组学分析表明,不平衡代谢通过ALE重组代谢扰动,诱导生长延迟和转录组及翻译组重构。上述研究说明我们对基因功能、代谢和基因组的认识还不全面,亟需对细菌基因组进行更全面的研究以填补我们知识空白,解答基因组缩减生物体所呈现的表型特征。
6 结语与展望
回顾全文,首先介绍了基于各种方法阐明必需基因集的研究,并依此设计最小(或缩减)基因组。通过比较多个基因组对必需基因进行计算研究,表明只有不到300个基因是细菌生存复制必需的。然而,这种比较低估了必需基因的数量,因为基因的功能可以被其他基因取代。利用转座子诱变、敲除、asRNA和CRISPRi技术进行实验评估,可揭示必需基因的直接和精确信息。但是,由于上述实验方法使基因破坏或失活,所以无法研究基因失活组合(如合成基因致死),需要开发更精细的方法以阐明基因之间相互关联的上位相互作用。
然后,介绍了以前构建的缩减基因组的特点及其构建方法。科学家们试图通过缩减已有基因组,使之成为最小基因组。现有的研究结果证明,缩减基因组是一把“双刃剑”,一方面,基因组缩减菌株可保留原始菌株优势特性,甚至还具有诸如更高的转化效率和生物化学法生产等有利特性;另一方面,仍会出现意想不到的表型,如生长迟缓和异常细胞形态等。这说明我们对基因组、基因功能和代谢的认识还不全面,还需要对细菌基因组进行更全面的研究以填补知识空白。
现今,高速发展的生物技术使设计基因网络,生物合成途径乃至整个基因组的构建成为可能,将遗传学和基因组学领域从描述性应用转移到了合成应用。在合成小型病毒基因组之后,DNA组装和重写技术的进步使细菌基因组(例如生殖支原体)的层次化合成成为可能[114],并且通过将密码子的数量从64个减少到57个来重新编码大肠杆菌基因组。目前,合成基因组学已经发展到合成整个真核基因组的地步。合成酵母基因组计划(Sc2.0)正在进行中,旨在重写所有16个酿酒酵母染色体;到2018年,已经设计并合成了6.5条染色体。使用自下而上的装配并应用全基因组的改变将增进对基因组结构和功能的理解。这种方法不仅将为真核染色体的系统研究提供平台,还将产生可能适用于医学和工业应用的各种“简化”菌株。Sc2.0的目的是设计和完全化学合成16条染色体,这些染色体包含来自啤酒酵母的1250万个碱基和一个带有所有tRNA基因的“新染色体”(http://syntheticyeast.org/)[115]。该项目不仅将为真核染色体的系统研究提供一个平台,还将通过其“从构建到理解”的过程扩展生物学知识的范围。
从小型病毒基因组的合成到酵母染色体Sc2.0,从罕见的密码子替代到全基因组重新编码,合成基因组学的可行性和能力已得到一次又一次的证明。Sc2.0着重介绍了用于生产非天然药物和工业化合物(如青蒿素)的酵母“底盘”的开发。基于合成染色体,可以将多种异源途径直接整合到酵母基因组中。合成基因组学还使改组基因组以快速生成新的基因组并筛选所需的特性成为可能。
最后,合成最小基因组是合成基因组学领域的一个里程碑,高通量基因合成技术正在持续发展过程中,凭借其已被证实的优势,使得最小基因组生物成为科学、工业和许多其他应用的理想生物平台,相信随着系统和合成生物学前沿技术的不断发展,终将解决目前存在的许多障碍和挑战。
[1] Bullmore E, Sporns O. The economy of brain network organization., 2012, 13(5): 336–349.
[2] Felleman DJ, Van Essen DC. Distributed hierarchical processing in the primate cerebral cortex., 1991, 1(1): 1–47.
[3] Cox DD, Dean T. Neural networks and neuroscience- inspired computer vision., 2014, 24(18): R921–R929.
[4] Krizhevsky A, Sutskever I, Hinton GE. ImageNet classification with deep convolutional neural networks., 2017, 60(6): 84–90.
[5] Maass W. Networks of spiking neurons: the third generation of neural network models., 1997, 10(9): 1659–1671.
[6] Mcculloch WS, Pitts W. A logical calculus of the ideas immanent in nervous activity., 1990, 52(1–2): 99–115.
[7] Nair V, Hinton GE. Rectified linear units improve restricted boltzmann machines vinod nair., 2010, 807–814.
[8] Rumelhart DE, Hinton GE, Williams RJ. Learning representations by back-propagating errors., 1986, 323(6088): 533–536.
[9] Izhikevich EM. Simple model of spiking neurons., 2003, 14(6): 1569–1572.
[10] McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria., 2011, 10(1): 13–26.
[11] Moran NA. Accelerated evolution and Muller's rachet in endosymbiotic bacteria., 1996, 93(7): 2873–2878.
[12] Mira A, Ochman H, Moran NA. Deletional bias and the evolution of bacterial genomes., 2001, 17(10): 589–596.
[13] Nilsson AI, Koskiniemi S, Eriksson S, Kugelberg E, Hinton JCD, Andersson DI. Bacterial genome size reduction by experimental evolution., 2005, 102(34): 12112–12116.
[14] Hebb DO. The Organization of behavior: a neuropsychological theory. John Wiley, Chapman & Hall, 2013.
[15] Mccutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria., 2011, 10(1): 13–26.
[16] Moran NA, Mira A. The process of genome shrinkage in the obligate symbiont Buchnera aphidicola., 2001, 2(12): h51–h54.
[17] Bennett GM, Moran NA. Small, smaller, smallest: the origins and evolution of ancient dual symbioses in a phloem-feeding insect., 2013, 5(9): 1675–1688.
[18] Mao C, Labean TH, Relf JH, Seeman NC. Logical computation using algorithmic self-assembly of DNA triple-crossover molecules., 2000, 407(6803): 493–496.
[19] Seelig G, Soloveichik D, Zhang DY, Winfree E. Enzyme-free nucleic acid logic circuits., 2006, 314(5805): 1585–1588.
[20] Zhang BT, Jang H. Molecular programming: evolving genetic programs in a test tube., 2005, 1761–1768.
[21] Zhang BT, Kim JK. DNA hypernetworks for information storage and retrieval[C]. 12th International Meeting on DNA Computing, 2006, 298–307.
[22] Benenson Y, Paz-Elizur T, Adar R, Keinan E, Livneh Z, Shapiro E. Programmable and autonomous computing machine made of biomolecules.,2001, 414(6862): 430–434.
[23] Yurke B, Turberfield AJ, Mills Jr AP, Simmel FC, Neumann JL. A DNA-fuelled molecular machine made of DNA., 2000, 406(6796): 605–608.
[24] Stojanovic MN, Stefanovic D. A deoxyribozyme-based molecular automaton., 2003, 21(9): 1069–1074.
[25] Pei RJ, Matamoros E, Liu MH, Stefanovic D, Stojanovic MN. Training a molecular automaton to play a game., 2010, 5(11): 773–777.
[26] Katz E, Privman V. Enzyme-based logic systems for information processing., 2010, 39(5): 1835–1857.
[27] Abbott LF, Nelson SB. Synaptic plasticity: taming the beast., 2000, 3 Suppl.: 1178–1183.
[28] Lichtsteiner P, Posch C, Delbruck T. A 128× 128 120 dB 15 μs latency asynchronous temporal contrast vision sensor., 2008, 43(2): 566–576.
[29] Liu SC, Delbruck T. Neuromorphic sensory systems., 2010, 20(3): 288–295.
[30] Maher MAC, Deweerth SP, Mahowald MA, Mead CA. Implementing neural architectures using analog vlsi circuits., 1989, 36: 643–652.
[31] Mead C. Neuromorphic electronic systems., 1990, 78(10): 1629–1636.
[32] Mead CA. Neural hardware for vision., 1987, 50(5).
[33] Huang YY, He L. DNA computing research progress and application. 2011 6th International Conference on Computer Science & Education (ICCSE), 2011, 232– 235.
[34] Seeman NC. DNA in a material world., 2003, 421(6921): 427–431.
[35] Steele G, Stojkovic V. Agent-oriented approach to DNA computing. 2004 IEEE Computational Systems Bioinformatics Conference, 2004.
[36] Greengard S. Cracking the code on DNA storage., 2017, 60(7): 16–18.
[37] Heckel R, Shomorony I, Ramchandran K, Tse DNC. Fundamental limits of DNA storage systems. 2017.
[38] Ghatak S, King ZA, Sastry A, Palsson BO. The y-ome defines the 35% of Escherichia coli genes that lack experimental evidence of function., 2019, 47(5): 2446–2454.
[39] Riley M, Abe T, Arnaud MB, Berlyn MKB, Blattner FR, Chaudhuri RR, Glasner JD, Horiuchi T, Keseler IM, Kosuge T, Mori H, Perna NT, Plunkett G, Rudd KE, Serres MH, Thomas GH, Thomson NR, Wishart D, Wanner BL. Escherichia coli K-12: a cooperatively developed annotation snapshot--2005., 2006, 34(1): 1–9.
[40] Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection., 2006, 2: 2006.0008.
[41] Moya A, Gil R, Latorre A, Peretó J, Garcillán-Barcia MP, de la Cruz F. Toward minimal bacterial cells: evolution vs. design., 2009, 33(1): 225–235.
[42] Williams KP, Sobral BW, Dickerman AW. A robust species tree for the Alphaproteobacteria., 2007, 189(13): 4578–4586.
[43] Gray MW. Mitochondrial evolution., 2012, 4(9): a11403.
[44] Boussau B, Karlberg EO, Frank AC, Legault BA, Andersson SGE. Computational inference of scenarios for α-proteobacterial genome evolution., 2004, 101(26): 9722–9727.
[45] Mizoguchi H, Sawano Y, Kato JI, Mori H. Superpositioning of deletions promotes growth ofwith a reduced genome., 2008, 15(5): 277–284.
[46] Stewart GC. Taking shape: control of bacterial cell wall biosynthesis., 2005, 57(5): 1177–1181.
[47] Silverman DJ, Wisseman Jr CL, Waddell A.studies of Rickettsia-host cell interactions: ultrastructural study of Rickettsia prowazekii-infected chicken embryo fibroblasts., 1980, 29(2): 778–790.
[48] Tully JG, Taylor-Robinson D, Cole RM, Rose DL. A newly discovered mycoplasma in the human urogenital tract., 1981, 1(8233): 1288–1291.
[49] Schröder D, Deppisch H, Obermayer M, Krohne G, Stackebrandt E, Hölldobler B, Goebel W, Gross R. Intracellular endosymbiotic bacteria of Camponotus species (carpenter ants): systematics, evolution and ultrastructural characterization., 1996, 21(3): 479–489.
[50] Aksoy S. Wigglesworthia gen. nov. and Wigglesworthia glossinidia sp. nov., Taxa consisting of the mycetocyte- associated, primary endosymbionts of tsetse flies., 1995, 45(4): 848–851.
[51] Moran NA, Dale C, Dunbar H, Smith WA, Ochman H. Intracellular symbionts of sharpshooters (insecta: hemiptera: cicadellinae) form a distinct clade with a small genome., 2003, 5(2): 116–126.
[52] Griffiths GW, Beck SD. Effects of antibiotics on intracellular symbiotes in the pea aphid, Acyrthosiphon pisum., 1974, 148(3): 287–300.
[53] von Dohlen CD, Kohler S, Alsop ST, McManus WR. Mealybug beta-proteobacterial endosymbionts contain gamma-proteobacterial symbionts., 2001, 412(6845): 433–436.
[54] Kopylova NV, Mikhaĭlova ZM, Fokina TV, Rybakova EP. Dynamics of the humoral immunity indices in children with phenylketonuria against a background of diet therapy and after its discontinuance., 1979, (6): 57–59.
[55] Oike M, Kitamura K, Kuriyama H. Histamine H3-receptor activation augments voltage-dependent Ca2+current via GTP hydrolysis in rabbit saphenous artery., 1992, 448: 133–152.
[56] Zarrin F, Bornhop DJ, Dovichi NJ. Laser Doppler velocimetry for particle size determination by light scatter within the sheath flow cuvette., 1987, 59(6): 854–860.
[57] Nakabachi A, Yamashita A, Toh H, Ishikawa H, Dunbar H E, Moran NA, Hattori M. The 160-Kilobase genome of the bacterial endosymbiont carsonella., 2006, 314(5797): 267.
[58] Ashby J. SCE induction by CCNU and pregnancy., 1989, 222(4): 299.
[59] Mushegian AR, Koonin EV. A minimal gene set for cellular life derived by comparison of complete bacterial genomes., 1996, 93(19): 10268–10273.
[60] Brown JR, Douady CJ, Italia MJ, Marshall WE, Stanhope MJ. Universal trees based on large combined protein sequence data sets., 2001, 28(3): 281–285.
[61] Harris JK, Kelley ST, Spiegelman GB, Pace NR. The genetic core of the universal ancestor., 2003, 13(3): 407–412.
[62] Koonin EV. Comparative genomics, minimal gene-sets and the last universal common ancestor., 2003, 1(2): 127–136.
[63] Charlebois RL, Doolittle WF. Computing prokaryotic gene ubiquity: rescuing the core from extinction., 2004, 14(12): 2469–2477.
[64] Hutchison CA, Peterson SN, Gill SR, Cline RT, White O, Fraser CM, Smith HO, Venter JC. Global transposon mutagenesis and a minimal Mycoplasma genome., 1999, 286(5447): 2165–2169.
[65] Itaya M. An estimation of minimal genome size required for life., 1995, 362(3): 257–260.
[66] Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, Arnaud M, Asai K, Ashikaga S, Aymerich S, Bessieres P, Boland F, Brignell SC, Bron S, Bunai K, Chapuis J, Christiansen LC, Danchin A, Débarbouille M, Dervyn E, Deuerling E, Devine K, Devine SK, Dreesen O, Errington J, Fillinger S, Foster SJ, Fujita Y, Galizzi A, Gardan R, Eschevins C, Fukushima T, Haga K, Harwood CR, Hecker M, Hosoya D, Hullo MF, Kakeshita H, Karamata D, Kasahara Y, Kawamura F, Koga K, Koski P, Kuwana R, Imamura D, Ishimaru M, Ishikawa S, Ishio I, Le Coq D, Masson A, Mauël C, Meima R, Mellado RP, Moir A, Moriya S, Nagakawa E, Nanamiya H, Nakai S, Nygaard P, Ogura M, Ohanan T, O'Reilly M, O'Rourke M, Pragai Z, Pooley HM, Rapoport G, Rawlins JP, Rivas LA, Rivolta C, Sadaie A, Sadaie Y, Sarvas M, Sato T, Saxild HH, Scanlan E, Schumann W, Seegers JFML, Sekiguchi J, Sekowska A, Séror SJ, Simon M, Stragier P, Studer R, Takamatsu H, Tanaka T, Takeuchi M, Thomaides HB, Vagner V, van Dijl JM, Watabe K, Wipat A, Yamamoto H, Yamamoto M, Yamamoto Y, Yamane K, Yata K, Yoshida K, Yoshikawa H, Zuber U, Ogasawara N. Essential Bacillus subtilis genes., 2003, 100(8): 4678–4683.
[67] Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection., 2006, 2: 2006.0008.
[68] Glass JI, Assad-Garcia N, Alperovich N, Yooseph S, Lewis MR, Maruf M, Hutchison CA, Smith HO, Venter JC. Essential genes of a minimal bacterium., 2006, 103(2): 425–430.
[69] Hutchison CA, Peterson SN, Gill SR, Cline RT, White O, Fraser CM, Smith HO, Venter JC. Global transposon mutagenesis and a minimal Mycoplasma genome., 1999, 286(5447): 2165–2169.
[70] Ji Y, Zhang B, Van SF, Warren HP, Woodnutt G, Burnham MK, Rosenberg M. Identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA., 2001, 293(5538): 2266–2269.
[71] Gerdes SY, Scholle MD, Campbell JW, Balázsi G, Ravasz E, Daugherty MD, Somera AL, Kyrpides NC, Anderson I, Gelfand MS, Bhattacharya A, Kapatral V, D'Souza M, Baev MV, Grechkin Y, Mseeh F, Fonstein MY, Overbeek R, Barabási AL, Oltvai ZN, Osterman AL. Experimental determination and system level analysis of essential genes inMG1655., 2003, 185(19): 5673–5684.
[72] Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression., 2013, 152(5): 1173–1183.
[73] Rousset F, Cui L, Siouve E, Becavin C, Depardieu F, Bikard D, Blokesch M. Genome-wide CRISPR-dCas9 screens in E. coli identify essential genes and phage host factors., 2018, 14(11): e1007749.
[74] Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells., 2014, 343(6166): 84–87.
[75] Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system., 2014, 343(6166): 80–84.
[76] Yu BJ, Kim C. Minimization of the Escherichia coli genome using the Tn5-targeted Cre/loxP excision system., 2008, 416: 261–277.
[77] Nijman SMB. Synthetic lethality: General principles, utility and detection using genetic screens in human cells., 2011, 585(1): 1–6.
[78] Kolisnychenko V, Plunkett G, Herring CD, Fehér T, Pósfai J, Blattner FR, Pósfai G. Engineering a reducedgenome., 2015, 12(4): 640–647.
[79] Pósfai G, Plunkett G, Fehér T, Frisch D, Keil GM, Umenhoffer K, Kolisnychenko V, Stahl B, Sharma SS, de Arruda M, Burland V, Harcum SW, Blattner FR. Emergent properties of reduced-genome Escherichia., 2006, 312(5776): 1044–1046.
[80] Lee JH, Sung BH, Kim MS, Blattner FR, Yoon BH, Kim JH, Kim SC. Metabolic engineering of a reduced- genome strain offor L-threonine production., 2009, 8: 2.
[81] Park MK, Lee SH, Yang KS, Jung SC, Lee JH, Kim SC. Enhancing recombinant protein production with an Escherichia coli host strain lacking insertion sequences., 2014, 98(15): 6701–6713.
[82] Hashimoto M, Ichimura T, Mizoguchi H, Tanaka K, Fujimitsu K, Keyamura K, Ote T, Yamakawa T, Yamazaki Y, Mori H, Katayama T, Kato JI. Cell size and nucleoid organization of engineeredcells with a reduced genome., 2005, 55(1): 137–149.
[83] Westers H, Dorenbos R, van Dijl JM, Kabel J, Flanagan T, Devine KM, Jude F, Seror SJ, Beekman AC, Darmon E, Eschevins C, de Jong A, Bron S, Kuipers OP, Albertini AM, Antelmann H, Hecker M, Zamboni N, Sauer U, Bruand C, Ehrlich DS, Alonso JC, Salas M, Quax WJ. Genome engineering reveals large dispensable regions in bacillus subtilis., 2003, 20(12): 2076–2090.
[84] Biswas I, Gruss A, Ehrlich SD, Maguin E. High- efficiency gene inactivation and replacement system for gram-positive bacteria., 1993, 175(11): 3628–3635.
[85] Choe D, Lee JH, Yoo M, Hwang S, Sung BH, Cho S, Palsson B, Kim SC, Cho BK. Adaptive laboratory evolution of a genome-reduced., 2019, 10(1): 935.
[86] Reuβ DR, Altenbuchner J, Mäder U, Rath H, Ischebeck T, Sappa PK, Thürmer A, Guérin C, Nicolas P, Steil L, Zhu BY, Feussner I, Klumpp S, Daniel R, Commichau FM, Völker U, Stülke J. Large-scale reduction of the Bacillus subtilis genome: consequences for the transcriptional network, resource allocation, and metabolism., 2017, 27(2): 289–299.
[87] Ara K, Ozaki K, Nakamura K, Yamane K, Sekiguchi J, Ogasawara N. Bacillus minimum genome factory: effective utilization of microbial genome information., 2007, 46(Pt 3): 169–178.
[88] Morimoto T, Kadoya R, Endo K, Tohata M, Sawada K, Liu SG, Ozawa T, Kodama T, Kakeshita H, Kageyama Y, Manabe K, Kanaya S, Ara K, Ozaki K, Ogasawara N. Enhanced recombinant protein productivity by genome reduction in bacillus subtilis., 2008, 15(2): 73–81.
[89] Komatsu M, Uchiyama T, Omura S, Cane DE, Ikeda H. Genome-minimized Streptomyces host for the heterologous expression of secondary metabolism., 2010, 107(6): 2646–2651.
[90] Giga-Hama Y, Tohda H, Takegawa K, Kumagai H. Schizosaccharomyces pombe minimum genome factory., 2007, 46(Pt 3): 147–155.
[91] Gibson DG, Glass JI, Lartigue C, Noskov VN, Chuang RY, Algire MA, Benders GA, Montague MG, Ma L, Moodie MM, Merryman C, Vashee S, Krishnakumar R, Assad-Garcia N, Andrews-Pfannkoch C, Denisova EA, Young L, Qi ZQ, Segall-Shapiro TH, Calvey CH, Parmar PP, Hutchison CA, Smith HO, Venter JC. Creation of a bacterial cell controlled by a chemically synthesized genome., 2010, 329(5987): 52–56.
[92] Hutchison CA, Chuang RY, Noskov VN, Assad-Garcia N, Deerinck TJ, Ellisman MH, Gill J, Kannan K, Karas BJ, Ma L, Pelletier JF, Qi ZQ, Richter RA, Strychalski EA, Sun LJ, Suzuki Y, Tsvetanova B, Wise KS, Smith HO, Glass JI, Merryman C, Gibson DG, Venter JC. Design and synthesis of a minimal bacterial genome., 2016, 351(6280): aad6253.
[93] Sleator RD. The story of Mycoplasma mycoides JCVI-syn1.0: the forty million dollars microbe., 2010, 1(4): 229–230.
[94] Lajoie MJ, Kosuri S, Mosberg JA, Gregg CJ, Zhang D, Church GM. Probing the limits of genetic recoding in essential genes., 2013, 342(6156): 361–363.
[95] Fredens J, Wang KH, de la Torre D, Funke LFH, Robertson WE, Christova Y, Chia T, Schmied WH, Dunkelmann DL, Beránek V, Uttamapinant C, Llamazares AG, Elliott TS, Chin JW. Total synthesis ofwith a recoded genome., 2019, 569(7757):514–518.
[96] Benosman R, Ieng SH, Clercq C, Bartolozzi C, Srinivasan M. Asynchronous frameless event-based optical flow., 2012, 27: 32–37.
[97] Wongsuphasawat K, Gotz D. Exploring flow, factors, and outcomes of temporal event sequences with the outflow visualization., 2012, 18(12): 2659–2668.
[98] Rogister P, Benosman R, Ieng SH, Lichtsteiner P, Delbruck T. Asynchronous event-based binocular stereo matching., 2012, 23(2): 347–353.
[99] Osswald M, Ieng SH, Benosman R, Indiveri G. A spiking neural network model of 3D perception for event-based neuromorphic stereo vision systems., 2017, 7: 40703.
[100] Hinton GE, Srivastava N, Krizhevsky A, Sutskever I, Salakhutdinov RR. Improving neural networks by preventing co-adaptation of feature detectors., 2012, 3(4): 212–223.
[101] Deng J, Dong W, Socher R, Li LJ, Li FF. ImageNet: a large-scale hierarchical image database[C]. 2009 IEEE Conference on Computer Vision and Pattern Recognition, 2009, 248–255
[102] Van Rullen R, Thorpe SJ. Rate coding versus temporal order coding: what the retinal ganglion cells tell the visual cortex., 2001, 13(6): 1255–1283.
[103] Mao C, Labean TH, Relf JH, Seeman NC. Logical computation using algorithmic self-assembly of DNA triple-crossover molecules., 2000, 407(6803): 493–496.
[104] Hu YH, Liu HJ, Pfeiffer M, Delbruck T. DVS benchmark datasets for object tracking, action recognition, and object recognition., 2016, 10: 405.
[105] Qian JF, Ferguson TM, Shinde DN, Ramírez-Borrero AJ, Hintze A, Adami C, Niemz A. Sequence dependence of isothermal DNA amplification via EXPAR., 2012, 40(11): e87.
[106] Geiger A, Lenz P, Stiller C, Urtasun R. Vision meets robotics: The KITTI dataset., 2013, 32(11): 1231–1237.
[107] Barranco F, Fermuller C, Aloimonos Y, Delbruck T. A dataset for visual navigation with neuromorphic methods., 2016, 10: 49.
[108] Adleman LM. Molecular computation of solutions to combinatorial problems., 1994, 266(5187): 1021–1024.
[109] Ogihara M, Ray A. DNA-based parallel computation by “counting”., 1997, 255–264.
[110] Deaton R, Murphy RC, Rose JA, Garzon M, Franceschetti DR, Stevens SE. A DNA based implementation of an evolutionary search for good encodings for DNA computation. 1997.
[111] Parker J. Computing with DNA - although DNA clearly outclasses any silicon-based computer when it comes to information storage and processing speed, a DNA-based PC is still a long way off., 2003, 4(1): 7–10.
[112] Arkin A. Setting the standard in synthetic biology., 2008, 26(7): 771–774.
[113] Seeman N, Wang H, Liu B, Qi J, Li X, Yang X, Liu F, Sun WQ, Shen ZY, Wang Y, Sha RJ, Mao C, Zhang S, Fu TJ, Du SM, Mueller JE, Zhang Y, Chen J. The perils of polynucleotides: The experimental gap between the design and assembly of unusual DNA structures. 1998.
[114] LIANG QF, WANG Q, QI QS. Synthetic biology and rearrangements of microbial genetic material., 2011, 33(10): 1102–1112.梁泉峰,王倩,祁庆生. 合成生物学与微生物遗传物质的重构. 遗传, 2011, 33(10): 1102–1112.
[115] Xu HM, Xie ZX, Liu D, Wu Y, Li BZ, Yuan YJ. Design and synthesis of yeast chromosomes., 2017, 39(10): 865–876.徐赫鸣, 谢泽雄, 刘夺, 吴毅, 李炳志, 元英进. 酿酒酵母染色体设计与合成研究进展. 遗传, 2017, 39(10): 865–876.
Research progress of bacterial minimal genome
Jinyu Li1,2,3, Shan Yang2,3, Yujun Cui2,3, Tao Wang1, Yue Teng2,3
,,,,,,,,
Bacteria with the smallest genome contain genes necessary for self-sustaining replication only, giving the organisms advantages to serve as a potential industrial production platform. Many strains with reduced genomes have been constructed, owing to the development of high-throughput DNA sequencing and synthesis technology. This review first describes the concept of minimal genomes, summarizes the relevant research progress of bacterial essential genes, then systematically lists the work related to artificial reduction and synthesis of bacterial genomes, finally discusses the technical obstacles and limitations encountered in the process of designing and constructing reduced genomes, hoping to provide a theoretical basis for the experiment and application of artificially synthesized genomes.
bacteria; minimal genome; essential genes; synthetic biology; artificial design
2020-11-23;
2021-01-04
国家科技重大专项(编号:2018ZX10101003-002-011)和“艾滋病和病毒性肝炎等重大传染病防治”科技重大专项(编号:2018ZX10712- 001-003)资助[Supported by the National Science and Technology Major Project (No. 2018ZX10101003-002-011) , and the Major Infectious Diseases Such as AIDS and Viral Hepatitis Prevention and Control Technology Major Projects (No. 2018ZX10712001-003)]
李金玉,在读硕士研究生,专业方向:生物学。E-mail: jyli1996@163.com
滕越,博士,副研究员,研究方向:合成生物学。E-mail: yueteng@sklpb.org
10.16288/j.yczz.20-301
2021/2/1 15:12:50
URI: https://kns.cnki.net/kcms/detail/11.1913.R.20210201.1343.010.html
(责任编委: 谢建平)
猜你喜欢
林业科学(2022年1期)2022-03-23
今日农业(2021年11期)2021-08-13
中国蜂业(2021年5期)2021-05-22
生物学通报(2020年11期)2020-10-22
中成药(2018年7期)2018-08-04
浙江农林大学学报(2016年6期)2016-12-12
遗传(2014年3期)2014-02-28
世界科学(2014年8期)2014-02-28
华东理工大学学报(自然科学版)(2014年5期)2014-02-27
茶叶通讯(2014年4期)2014-02-27
