
中国彝族人群p表型家系分析及新A4GALT等位基因鉴定*
2021-02-24 00:20:06贺坤华苏品璨许先国林裕翔徐路琼徐银霞赵瑜马丽琼
临床输血与检验 2021年1期
贺坤华 苏品璨 许先国 林裕翔 徐路琼 徐银霞 赵瑜 马丽琼
2011年国际输血协会(ISBT)已将P血型系统重新命名为P1PK血型系统(编号003),P1PK血型系统包括Pl(003001)、PK(003002)及NOR抗原(003004);Globside血型系统仅有P抗原(028001);LKE抗原(209003)、PX2抗原(209004)则归入GLOB血型集合,也称糖苷脂集合[1-3]。p表型表现为患者红细胞上P、P1和PK、LKE这4种抗原全部缺失(null),因此其极易产生抗体[4,5]。
p表型在中国人群中极为罕见,在中国香港筛选100万人才发现1例[6]。目前国内有10余例的p表型报道,但对其分子机制研究甚少[7]。研究显示A4GALT编码的酶催化合成P1和PK抗原,B3GALNT1控制P抗原,A4GALT基因第3外显子发生变异导致氨基酸改变时,可能会引起α1,4-半乳糖基转移酶失活,导致P1、PK及其下游 P、LKE抗原丢失形成p表型[8]。
我们通过对1个p表型彝族家系的血清学和相关的A4GALT和B3GALNT1基因进行研究,发现了一个导致p表型的新的A4GALTc.456-457insACACCCC等位基因,阐明p表型形成的分子遗传机制,现报告如下。
材料与方法
1 标本来源 患者(先证者),女,25岁,孕12周,彝族,无输血史,云南曲靖人。患者在第一胎孕8周时自然流产,反复检查均未查明流产原因,现第2次怀孕来我院产检,做血型鉴定时发现ABO血型正反定型不一致,丈夫为汉族,其余家系成员为彝族,均为云南曲靖人。经医院伦理委员会审核通过并签署知情同意书后,采集患者及家系成员血样,EDTA-K2抗凝管和促凝管各5 mL,用于血清学检测和基因检测。
2 仪器和试剂 A B O D E血型检测卡(批号20190911)、Rh分型卡(批号20191017)、抗球蛋白检测卡(批号20191017)、ABO血型反定型试剂盒(批号20191007)、抗筛细胞(批号20190823)、2-Me(批号20190501)均由长春博迅生物技术有限责任公司生产;抗人球蛋白试剂(批号20190209)、抗-H(批号20190707),抗-P1(批号20190130)、抗-M(批号20191031)、抗-N(批号20191204),均由上海血液生物医药有限责任公司生产;谱细胞(批号20191020)、红细胞稀有血型基因分型试剂盒(批号20191109)、商用稀有血型测序试剂盒(批号20191018)均由江苏中济万泰生物医药有限公司生产;人源抗-Tja、人p表型红细胞均由浙江省血液中心提供;TD-A型血型血清学用离心机、FYQ型免疫微柱孵育器(长春博研科学仪器有限责任公司生产);KA-2200型离心机(日本久保田生产);SSW-600-2S型电热恒温水浴槽由上海博讯实业有限公司医疗设备厂生产。所有试剂均在有效期内。
3 血型鉴定、直接抗球蛋白试验、抗体筛查及鉴定均按说明书和文献介绍方法[9]进行。
4 家系调查 对先证者及父母、丈夫、同胞、表姐、姑母、侄儿等共34人进行家系调查。
5 分子生物学检测 红细胞稀有血型基因分型采用荧光定量PCR法;同时采用商用稀有血型测序试剂盒对A4GALT和B3GALNT1进行Sanger测序。A4GALT、B3GALNT1基因扩增和PCR产物纯化、PCR产物直接序列分析参照相关文献[4,7]。序列比对分析采用GENEWIZ软件,测序结果与A4GALT(Gen Bank AJ245581)和B3GALNT1(Gen Bank AB050855)参考序列进行比对,以确定单核苷酸多态性(single nucleotide polymorphisms,SNPs)。
结 果
1 血清学检查结果 先证者及其哥哥ABO正定型为B型,反定型发现其血清与A、B、O标准细胞反应均为阳性,怀疑有意外抗体。抗体鉴定发现先证者及其哥哥血清与筛选细胞和谱细胞反应均阳性且凝集强度一致,在盐水试验中均出现2+凝集、在抗人球介质中均出现3+s凝集,直抗和自身对照阴性,可与自身抗体区分,并怀疑有高频抗原抗体。进一步检测发现,该抗体能在室温反应5 min后或37℃温育后引起红细胞溶血,结合先证者的红细胞表型,可以排除抗-H、抗-I等IgM抗体的特异性;进一步鉴定发现,先证者及其哥哥血清与人p表型红细胞反应阴性;细胞与人源抗-PP1PK血清反应阴性,说明先证者及哥哥均是罕见的p表型,其血清中均含有能与所有非p表型红细胞反应的抗-PP1PK抗体。采用EDTA-2K抗凝血浆与抗筛细胞和谱细胞反应均有凝集反应,而无溶血反应,提示该抗体具有补体活性。血清经2-Me处理后盐水试验阴性,IAT试验阳性,说明该抗体具有IgM和IgG的性质,这些均符合抗-PP1PK特异性(血清学检查结果与昆明市血液中心和浙江省血液中心结果一致)。
2 先证者红细胞稀有血型基因分型结果 CCDee, LW(a+b-), Fy(a+b-), Jk(a+b-), MMSs Mur-,Di(a-b+), Wr(a-b+), kk, Kp(a-b+), Co(a+b-), Do(a+b-), Au(a+b+), Yt(a+b-)。
3 家系调查结果 先证者(27)及其哥哥(28)为罕见p表型,其他家系成员为正常的P1或P2表型(图1)。基因携带情况详见图注。
4A4GALT和B3GALNT1测序结果 测序结果显示(图2),先证者及其哥哥为A4GALT456-457insACACCCC(Bank IT:MG812384)纯合突变。同时在该家系中发现6例456-457insACACCCC杂合突变,7例109 A>G和987G>A杂合突变,3例IVS+228C>T杂合突变。其他家系成员A4GALT序列与正常参考序列(Gen Bank AJ245581)相同;所有家系成员B3GALNT1序列均与参考序列(Gen Bank AB050855)完全一致,即在该基因上未发现任何碱基突变。
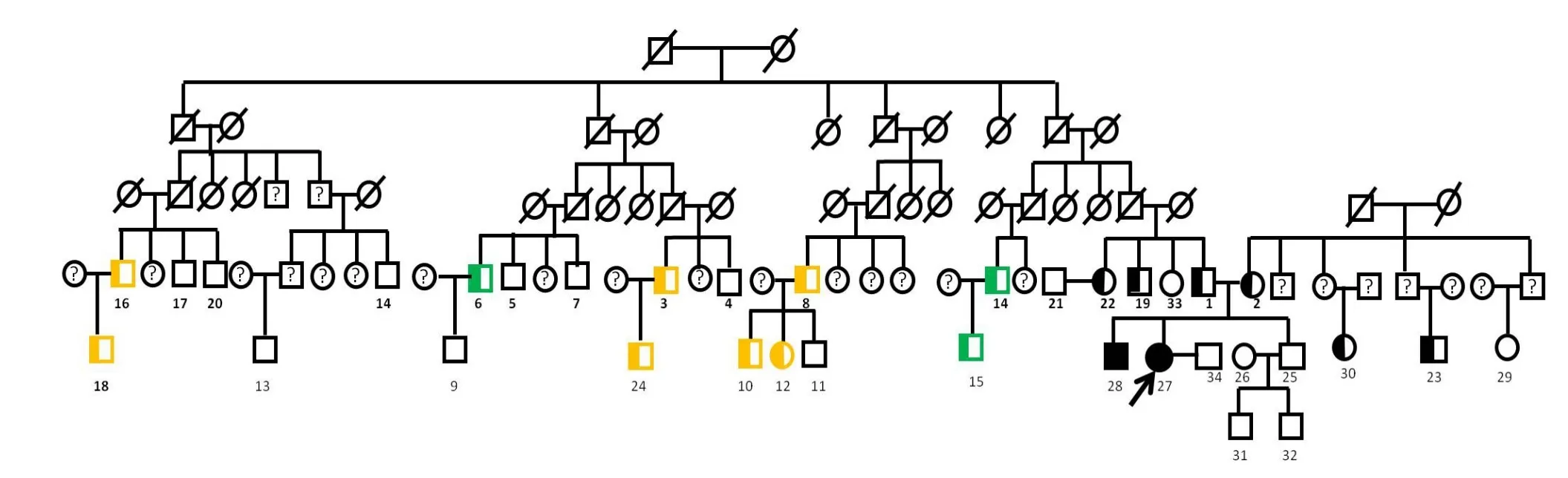
图1 先证者家系图
讨 论
研究表明A4GALT基因(也称CD77合成酶基因)突变可导致p表型[10,11]。A4GALT基因定位于22q11.2-q13.2,基因组全长为29500 bp,有3个外显子,其中外显子1长25 bp,外显子2长141 bp,外显子3长1876 bp,开始密码子和基因编码区都位于第3外显子上,编码353个氨基酸。目前国际上发现的与p表型相关的A4GALT基因突变已达到52个,大多数都发生在外显子3上[1],突变类型包括单核苷酸突变、核苷酸插入或缺失等[4]。
使用经典的血清学方法检测血型,必须依赖大量的标准抗血清,稀有血型抗血清难以获得且价格昂贵[12]。本研究中采用荧光定量PCR法进行红细胞稀有血型基因分型,可用1份DNA标本检测多个血型系统的多种抗原,也可检出稀有血型和辅助检测高频抗原抗体[13]。基因分型结果排除了常见的高频抗原抗体,包括Jknull、RH变异体、Fya-、Dib-、LWa-、Colton和Domcrock,但不排除JMH、Vel、Lan、Lutheran和p。进一步采用人p表型红细胞和人源抗-PP1PK血清等血清学试验进行验证,结果充分说明先证者及哥哥均是罕见的p表型。
家系基因测序发现,所有家系成员B3GALNT1序列均与正常参考序列相同,而先证者及其哥哥A4GALT基因编码区存在c.456-457insACACCCC纯合突变。我们将该序列在NCBI (National Center for Biotechnology Information, NCBI )基因库中进行Blast搜索,没有发现相同的等位基因;进一步与ISBT、ERYTHROGENE数据库中的数据进行比对,没有找到相同序列。已知文献中未发现关于该突变的报道,因此我们将c.456-457insACACCCC突变序列作为新的A4GALT等位基因上报NCBI数据库并取得登记号(Bank IT:MG812384)。c.456-457insACACCCC突变导致开放阅读框移码,从第156个氨基酸开始发生序列改变,终止于第283个氨基酸。突变基因翻译的氨基酸链与野生型(353个氨基酸)相比,只有前155个氨基酸是相同的,如此大的氨基酸序列改变将引起多肽链空间折叠构象的改变,进而引起酶活性丧失,导致酶催化产物P1和Pk抗原及其下游的P、LKE抗原不能正常合成而形成p表型。
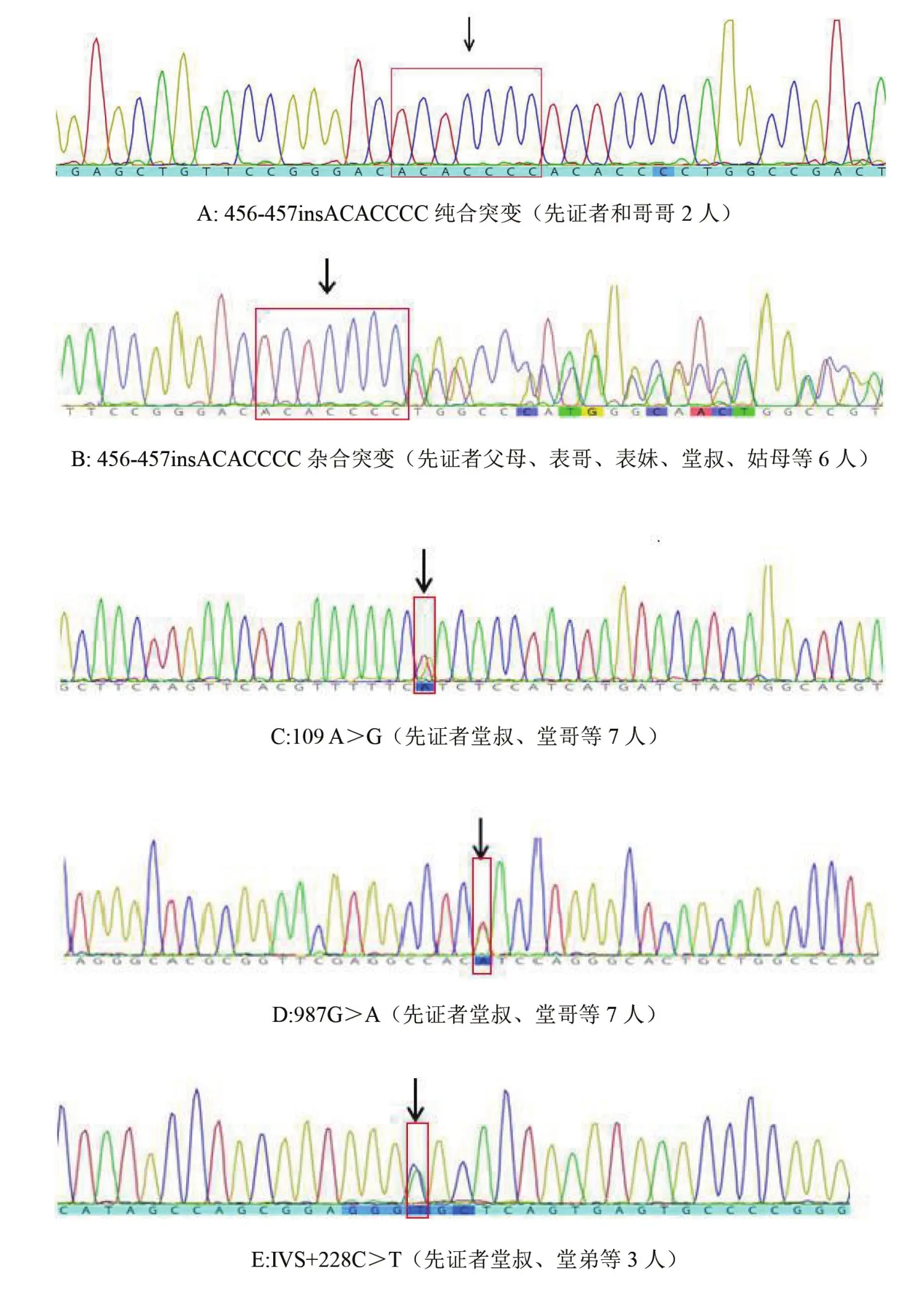
图2 A4GALT基因序列图谱(突变位点由红框和箭头标示)
家系调查发现,先证者及其哥哥均为c.456-457insACACCCC纯合突变,其父母均为c.456-457insACACCCC突变携带者,因而推测先证者及哥哥分别继承了父母的突变基因,从而导致p表型,另有4名家系成员也携带此突变,但所有c.456-457insACACCCC杂合子均表现为非p表型。此外该家系中检出的7例109A>G和987G>A杂合突变,3例IVS+228C>T杂合突变也未产生p表型,只有纯合子才表现罕见的p表型, 且p表型往往在同代中出现,表明其遗传方式为常染色体隐性遗传[14]。
对A4GALT基因中SNPs的系统研究表明[15],A4GALT基因的差异性表达,是P1/P2血型的分子遗传基础,其中P1个体中的A4GALT表达水平高于P2个体。同时有研究显示[16],单个碱基插入(A4GALT/insC)会降低A4GALT活性。至于109A>G和987G>A、IVS+228C>T突变是否会影响酶活性,目前尚未见相关文献报道。另外,多名家系成员同时携带109A>G和987G>A突变,而大部分家系成员同时不携带109A>G和987G>A突变,未检测到只携带其中一种突变的个体,因而推测109A>G和987G>A是发生在同一等位基因上的两种突变。鉴于实验条件限制,未进一步采用克隆测序进行证实。本研究从多名家系成员中检出多种A4GALT基因突变,说明该家系具有丰富的A4GALT基因多态性。因条件限制,本次未能深入进行体外表达及酶活性研究,然而从先证者表现为p表型的事实可以推测c.456-457insACACCCC突变可造成αl,4-半乳糖基转移酶活性丧失。综上所述,我们首次在彝族家系中发现的A4GALT基因c.456-457insACACCCC突变导致了p表型的形成。
据文献报道,云南第1例p表型是由济宁市中心血站耿微[5]发现的,患者系云南少数民族妇女,31岁,无输血史,孕2产2,因产后出血需要输血而进行血型鉴定和交叉配血时被发现。此后昆明市血液中心也在云南发现了1例p表型[17],系女性,24岁,佤族,为无偿献血者。云南是中国少数民族种类最多的省份,人口在6000人以上的世居少数民族有25个。全省少数民族人口数达1621.26万人,其中彝族是云南少数民族中人口最多的一个民族,主要分布在滇东北、滇中和滇北广大地区。由于民族迁徙和融合,云南彝族可能存在独特的遗传背景,进一步采用大样本研究其血型多态性,将有助于稀有血型资料库建立。(致谢:感谢台大附属医院输血科张志昇教授对本研究的指导和帮助!)
利益冲突 所有作者均声明不存在利益冲突
猜你喜欢
临床输血与检验(2022年3期)2022-06-22 02:52:50
中国生殖健康(2020年6期)2020-02-01 06:28:52
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
中国生殖健康(2018年6期)2018-11-06 07:09:30
现代园艺(2017年21期)2018-01-03 06:41:32
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
重庆医学(2015年12期)2015-03-05 05:52:54
现代检验医学杂志(2015年5期)2015-02-06 01:42:20
