
陆地棉酵母双杂交cDNA文库及VdSCP7诱饵载体的构建
2021-02-22 08:48韦春艳秦腾飞李玉青孙家良王清连
核农学报 2021年3期
韦春艳 秦腾飞 董 娜 李玉青 董 涛 郭 婷 孙家良 王清连
(1 河南科技学院/现代生物育种河南省协同创新中心/河南省棉麦分子生态与种质创新重点实验室,河南 新乡 453003; 2新疆农业大学农学院,新疆 乌鲁木齐 830000)
棉花黄萎病是一种土传维管束真菌病害,被称为棉花的“癌症”,严重影响棉花的产量和品质,对棉花生产造成了巨大的经济损失[1]。我国棉花黄萎病的主要致病菌为大丽轮枝菌(Verticillium dahlia)[2]。它是一种土传的半活体营养型丝状真菌,在世界各地分布广泛,可以侵染660 多种植物引发黄萎病[3],导致棉花、芥菜、马铃薯、甜菜、向日葵等许多重要经济作物连年发生病害[4-6]。由于大丽轮枝菌具有寄主范围广、类型多、变异快、存活时间久、抗逆性强等特点,致使棉花黄萎病难以防治[7-9]。且陆地棉抗性种质资源匮乏,抗性育种进展缓慢[5],因此研究棉花和黄萎病菌互作的分子机制,挖掘棉花抗病基因,对提高棉花抗病性具有重要意义。
酵母双杂交技术是研究蛋白之间互作的有效分子生物学方法[10],在检测蛋白之间互作和筛选未知蛋白方面得到了广泛的应用[11-13],构建高质量的cDNA 文库则是运用酵母双杂交技术的前提。王宇秋等[14]构建了水稻酵母双杂交cDNA 文库并利用文库筛选到了56 个与稻曲病菌效应因子Uv_1261 互作的蛋白。李帅等[15]构建了草莓酵母cDNA 文库筛选到15 个与草莓镶脉病毒P6 互作的寄主因子,为明确其互作机理提供了理论依据。瓮巧云等[16]筛选拟南芥cDNA 文库获得了4 个可能与抗灰霉病基因T1N6-22 互作的蛋白,为阐明其调控的分子机制奠定了基础。郑凯等[17]构建了海岛棉纤维均一化酵母cDNA 文库并筛选到了4 个与GbTCP5 互作的蛋白。还有研究者利用酵母双杂交筛选到了一些参与小麦逆境反应的候选蛋白[18-19]。但是,陆地棉酵母双杂交cDNA 文库的构建鲜见报道,近年来国内外学者对黄萎病的研究主要集中于抗病基因鉴定、病害循环、抗性品种培育等方面,并取得了一定的研究进展,但对于黄萎病菌和棉花互作机制的研究较少,棉花抗黄萎病机理还不十分清楚。通过构建棉花酵母双杂交cDNA 文库,研究黄萎病菌和棉花互作机理,可为提高棉花抗病性提供理论依据。研究表明,黄萎病菌效应因子可激活植物免疫反应提高植物抗病性[20],植株的免疫反应程度越强其抗病性也越强[21]。植物受病原体侵染时,可以通过模式识别受体(pattern recognition receptors, PRRs)识别病原体相关分子模式(pathogen-associated molecular patterns,PAMPs),从而放大先天性免疫,激活第一层防御反应(PAMP triggered immunity, PTI)[22]。病原菌要成功定植必须克服寄主的免疫防卫反应,所以病原菌又分泌效应蛋白进入寄主植物抑制其PTI 反应[23]。一些植物进化出了抗性蛋白能特异性识别病原菌效应因子触发另一层更强的免疫反应(effector-triggered immunity, ETI)来抑制病原菌的侵染[23-25]。大丽轮枝菌就可以分泌效应蛋白抑制宿主的防卫反应[26],轮枝菌属特异的外泌蛋白(VdSCP7)是一种靶向宿主细胞核的大丽轮枝菌效应蛋白,植物在大丽轮枝菌侵染过程中可以识别VdSCP7 并激活ETI 免疫反应,增强抗病性[27]。但是,目前关于VdSCP7 在棉花抗病过程中作用的分子机制尚不清楚。
本研究通过构建棉花cDNA 文库和诱饵载体pGBKT7-SCP7,从而利用酵母双杂交系统从棉花cDNA 文库中筛选与VdSCP7 互作的蛋白,为进一步从分子水平上解析VdSCP7 和棉花互作机理和提高棉花对黄萎病的抗性奠定基础,也为利用酵母双杂交系统研究黄萎病菌蛋白特别是病原菌效应因子与棉花互作提供资源。
1 材料与方法
1.1 主要材料
大丽轮枝菌Vd991 菌株由河南省棉麦分子生态与种质创新重点实验室保存;国审棉花品种百棉1 号由河南科技学院棉花研究所提供,26℃温室中培养,光照时间为14 h.d-1,相对湿度为60%~70%。
1.2 棉花总RNA 提取及ds cDNA 合成
取马铃薯葡萄糖琼脂培养基(potato dextrose agar,PDA)上活化培养的大丽轮枝菌菌丝,经完全培养基(complete medium,CM)液体培养基培养后过滤收集孢子,调整孢子浓度为1×107CFU.mL-1,用蘸根法[28]侵染生长2 周左右的棉花幼苗,孢子侵染根部后,分别于3、6、9、12 d 后取样,将棉花根系样品混合提取总RNA。具体步骤按照RNAiso Plus Reagent 试剂盒说明书(TaKaRa,日本)进行,1%的琼脂糖凝胶电泳检测RNA 完整性后,用Oligotex-dT30<Super>mRNA 纯化试剂盒(TaKaRa,日本)对mRNA 进行分离纯化,具体步骤参照说明书,用1%的琼脂糖凝胶电泳检测mRNA质量。再以含XhoⅠ酶切位点的Oligo(dT)18Anchor Primer 为引物,用SMARTerTMPCR cDNA 合成试剂盒(Clontech,美国)合成cDNA 第一链。
以含EcoR Ⅰ酶切位点的SMART (switching mechanism at 5′ end of RNA template)寡核苷酸为引物经long-distance PCR(LD-PCR)合成双链cDNA。双链cDNA 经EcoR Ⅰ和XhoⅠ酶切后,用CHROMA SPIN+TE-1000 (Clontech,美国) 柱纯化回收大于500 bp 的双链DNA(double stranded cDNA,ds cDNA),用于构建棉花cDNA 文库,具体步骤参照试剂盒说明书进行。
1.3 cDNA 文库的构建及扩增
将棉花ds cDNA 与pGADT7 质粒载体(Clontech,美国)用酶切连接的方法完成cDNA 文库的构建。将经EcoR Ⅰ和XhoⅠ酶切处理的pGADT7 载体回收产物与纯化回收的ds cDNA 用T4-DNA 连接酶连接。连接体系:7 μL pGADT7、10 μL ds cDNA、1 μL ddH2O、2 μL T4DNA 连接酶,16℃连接20 h。
cDNA 文库扩增:取连接产物加到大肠肝菌E.coliDH5α Electro-Cells(TaKaRa,日本)中,冰浴10 min 后转移到电击杯中,电转化条件:1.8 kV,200 Ω,25 μF,电转仪:E.colipulser(美国Bio-Rad 公司)。电击完成后立即加入1 mL 的SOC(super optimal broth with catabolite repression)液体培养基到转化产物中,随后转移到50 mL SOC 液体培养基中,37℃,250 r.min-1条件下复苏1 h,然后涂布于LB+Amp 抗性平板上,37℃倒置过夜培养后用LB 液体培养基收集所有菌体,将得到的菌体悬浮液于37℃、250 r.min-1震荡培养3 h左 右, 用Wizard ® Plus Maxipreps DNA purification System (Promega,美国)提取质粒完成cDNA 文库的扩增。
1.4 cDNA 文库质量和滴度检测
ds cDNA 和pGADT7 连接产物电转化到大肠杆菌后,取转化产物2 μL 分别稀释10 倍、100 倍后涂到LB+Amp 平板上,37℃培养过夜后,统计单克隆数计算原始文库容量。从平板上随机挑取32 个单克隆,以pGADT7-F/pGADT7-R 为引物(表1),通过PCR 扩增,用1%的琼脂糖凝胶电泳检测,鉴定文库平均插入片段的大小。取1.3 中震荡培养好的菌液100 μL,按10-2、10-4、10-63 个稀释度稀释后分别取100 μL 涂LB+Amp 平板,37℃培养过夜,统计平板上克隆数计算文库滴度(文库滴度:克隆数目/稀释混合物涂板体积×稀释倍数=CFU.mL-1)[29]。
1.5 pGBKT7-SCP7 诱饵表达载体构建
根据GenBank 中VdSCP7 基因序列(Gen ID:VDAG_07157)设计带有EcoR Ⅰ酶切位点的引物SCP7-F 与SCP7-R(表1)。液氮研磨大丽轮枝菌Vd991 菌丝体,用RNA 快速提取试剂盒提取大丽轮枝菌RNA,1%的琼脂糖电泳检测后;用反转录试剂盒获得大丽轮枝菌cDNA;纯化回收大丽轮枝菌cDNA 后。以Vd991 cDNA 为模板,SCP7-F 与SCP7-R 为引物扩增VdSCP7 基因。
用EcoR Ⅰ酶切并纯化回收载体pGBKT7,然后将VdSCP7 的PCR 回收产物用Assembly Mix 重组到pGBKT7 载体上,重组体系:0.5 μL pGBKT7、2 μLVdSCP7、2.5 μL 2×Assembly Mix,然后将重组产物转化到50 μL Trans1-T1 大肠杆菌感受态细胞中,转化步骤参考Trans1-T1 Phage Resistant 化学感受态细胞说明书(北京全式金生物技术有限公司),再提取大肠杆菌中pGBKT7-SCP7 质粒,经EcoR Ⅰ酶切并用1%琼脂糖凝胶电泳检测后,将诱饵载体pGBKT7-SCP7 送生工生物工程(上海)股份有限公司进行测序验证。
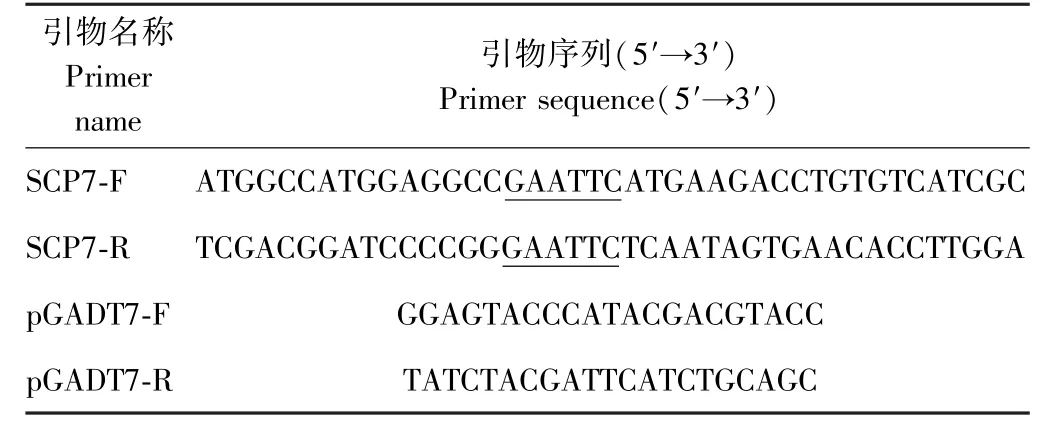
表1 引物信息Table 1 Primer information
1.6 pGBKT7-SCP7 诱饵载体自激活及毒性检测
参照Clontech 产品说明书,用LiAc 法制备酵母AH109感受态细胞(Clontech,美国)。将5 μL pGBKT7-SCP7 质粒、3 μL pGADT7 质粒、77 μL 无菌水和5 μL 热变性的Carrier DNA 加入酵母感受态细胞中混匀,再加入PEG/LiAc(240 μL 50%的PEG、36 μL 1 mol.L-1的LiAc)混匀,30℃、200 r.min-1震荡培养30 min 后,42℃热激30 min,12 000 r.min-1离心1 min,弃上清,用100 μL 0.9%NaCl 溶液重悬菌体。取适量菌液涂到SD/-Trp/-Leu 和SD/-Ade/-His/-Trp/-Leu 培养基(TaKaRa,日本)上,30℃培养3 d,观察结果,分析诱饵载体自激活活性。
将含pGBKT7-SCP7 和pGBKT7 空载体的酵母菌AH109,分别接种于SD/-Trp/Kan 的液体培养基中,30℃、250 r.min-1培养24 h,用722N 型分光光度计(上海精密仪器仪表有限公司)检测菌液OD600,鉴定诱饵蛋白对细胞的毒性。
2 结果与分析
2.1 棉花总RNA 提取及ds cDNA 的合成
提取被大丽轮枝菌侵染的棉花根系RNA,经测定RNA OD260/OD280为1.91,电泳检测,28S 和18S rRNA条带清晰,无拖尾,亮度比接近2 ∶1,说明总RNA 完整性较好,未发生降解(图1-A)。mRNA 纯化后呈弥散状分布,质量较高(图1-B)。合成的ds cDNA 于0.2~4.5 kb 之间呈弥散状,说明不同大小和丰度的mRNA 均有效反转录(图1-C)。ds cDNA 经酶切纯化去除小片段后,电泳检测条带仍呈弥散状,片段大小分布在0.5~3.0 kb 之间,纯化产物质量较高,可用于构建cDNA 文库(图1-D)。
2.2 文库质量和滴度检测
将纯化后的ds cDNA 连接到pGADT7 载体上,转入大肠杆菌中,涂布到平板上测定原始文库库容量大于2.0×106CFU。从平板上随机挑取32 个克隆,进行菌液PCR 检测,部分电泳图片如图2 所示,插入的cDNA 片段大小不一,文库多样性良好,插入片段平均长度大于1 kb,32 个克隆仅有2 个未检测到插入片段,重组率约94%。上述结果说明文库质量较高,可用于后续筛库试验。
取扩增后的文库菌悬液100 μL,按10-2、10-4、10-6稀释后分别取100 μL 涂LB+Amp 平板,37℃培养过夜,统计平板上克隆数计算文库滴度,最后得到扩增后的质粒文库滴度约2.0×109CFU.mL-1,满足试验要求所需的滴度。
2.3 诱饵载体(pGBKT7- SCP7)的构建
将测序结果正确的VdSCP7 重组至pGBKT7 载体上,并转化Trans1-T1 感受态细胞中,将转化产物涂布于LB+Kan 培养基上。挑取单克隆,摇菌培养,提取质粒,EcoR Ⅰ酶切pGBKT7-SCP7 重组质粒并电泳检测,切下的目的片段(即图3 中右图下方片段)与VdSCP7基因片段(651 bp)大小一致(图3),且与生工生物工程(上海)股份有限公司测序结果比对正确,说明诱饵载体pGBKT7-SCP7 构建成功。

图1 cDNA 文库的构建Fig.1 Construction of cDNA library
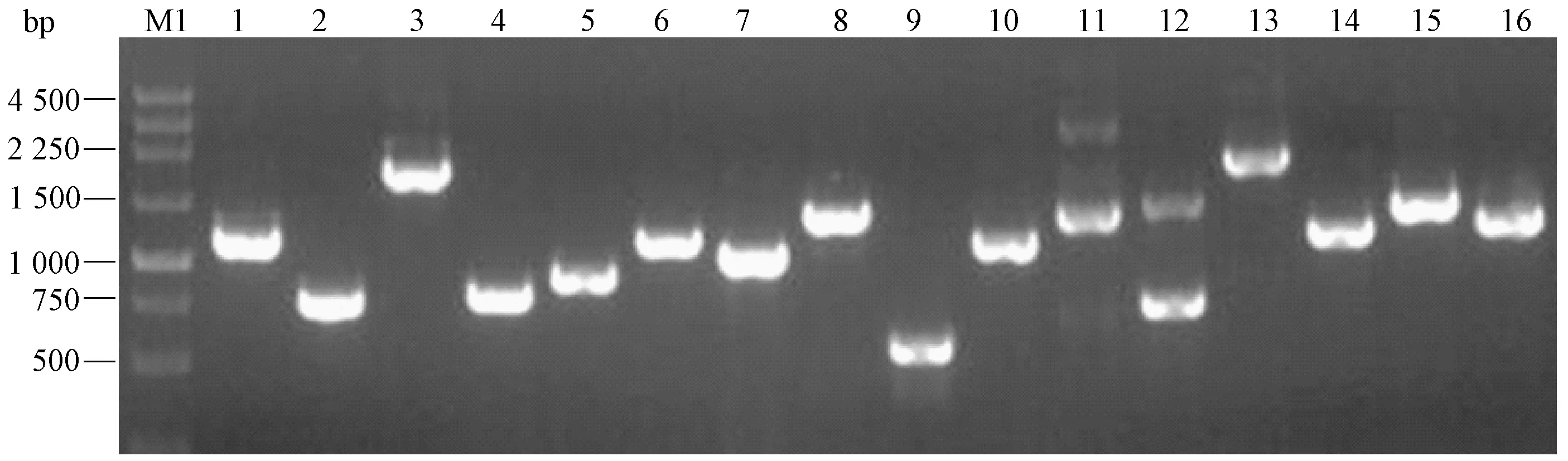
图2 PCR 检测cDNA 文库的插入片段Fig.2 PCR identification of inserts in the cDNA library

图3 诱饵载体(pGBKT7-SCP7)的构建Fig.3 Construction of bait vector pGBKT7-SCP7
2.4 诱饵载体pGBKT7-SCP7 自激活及毒性鉴定
含有pGBKT7-SCP7 和pGADT7 空载体的转化菌在SD/-Trp/-Leu 中正常生长(图4-A),在SD/-Ade/-His/-Trp/-Leu 中无生长(图4-B),说明诱饵载体pGBKT7-SCP7 无自激活活性。含pGBKT7-SCP7 和pGBKT7 空质粒的转化菌分别在SD/-Trp/Kan 液体培养基培养24 h 后,测定菌液OD600均大于0.8,说明诱饵载体pGBKT7-SCP7 对酵母细胞无毒性,可用于酵母双杂交系统进行下一步筛库试验。
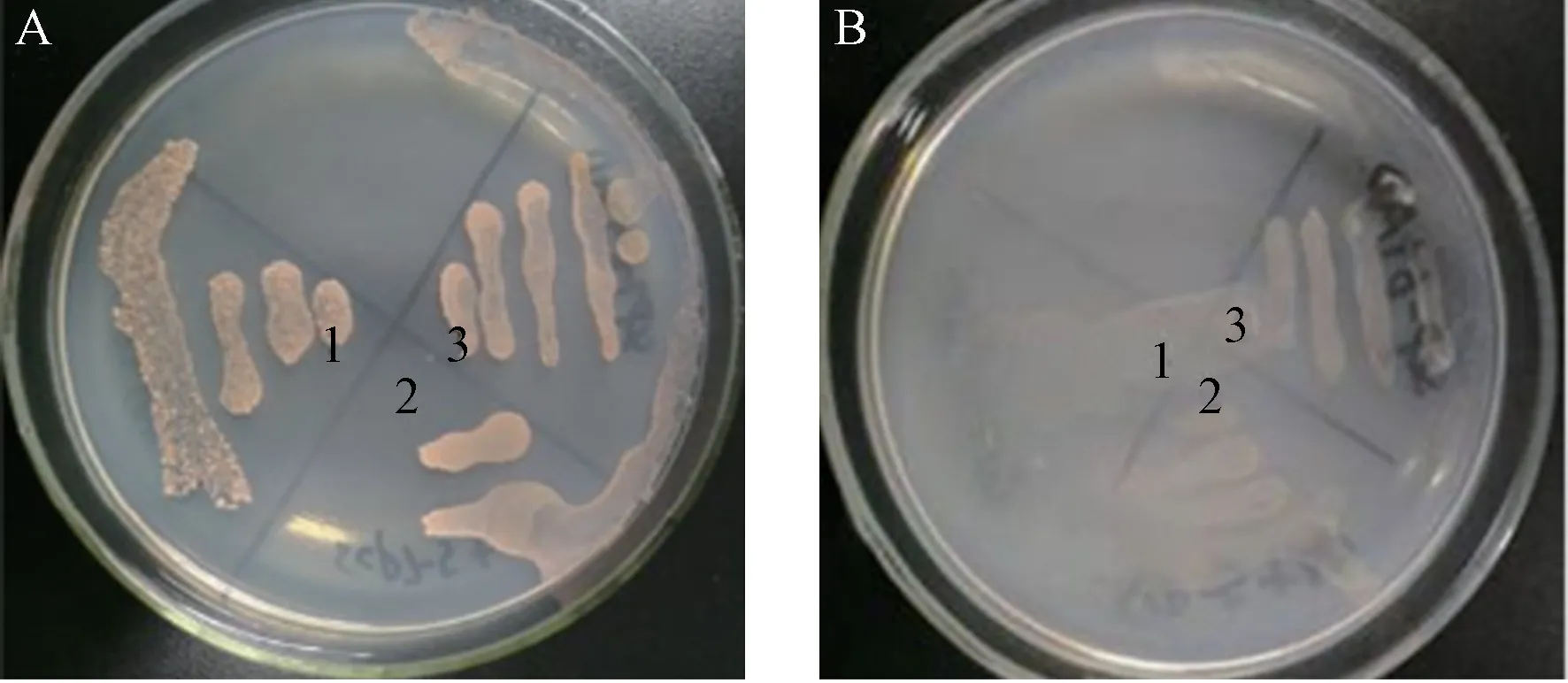
图4 pGBKT7-SCP7 的自激活验证Fig.4 Auto-activation identification of pGBKT7-SCP7
3 讨论
构建cDNA 文库是发掘新基因、研究基因功能的重要技术手段。已有研究构建了纤维发育、花发育、盐旱胁迫等相关的棉花cDNA 文库,并挖掘到许多与棉花生长发育和抗逆相关的关键基因[30]。Zhang 等[31]以海岛棉Pima90-53 为材料构建了cDNA 文库,得到了46 192 条高质量表达序列标签(expressed sequence tags,ESTs),获得了23 126 个unigenes,并发现了1 条新的和植物抗性相关的信号转导途径。马建辉等[32]构建了海岛棉cDNA 文库并筛选到了一系列在棉花花药发育中起重要作用的基因。Zhang 等[33]利用cDNA文库鉴定出了一些信号分子、转录因子等可能在棉花盐胁迫应答中起重要作用。张映霞等[34]和Zhang等[35]以黄萎病菌诱导处理后的陆地棉为材料构建cDNA 文库,得到了一些和抗病相关的基因。但这些棉花cDNA 文库都不能直接用于酵母双杂交系统进一步对筛到的关键基因进行分子机制的解析。通过构建棉花酵母双杂交cDNA 文库,筛选棉花中与黄萎病菌互作的蛋白,解析其互作的分子机理,对提高棉花抗病性至关重要。
酵母双杂交技术是研究蛋白质之间相互作用的高通量技术,已被广泛应用,且效率高,筛选量大[36],而高质量的cDNA 文库是运用酵母双杂交系统进行筛选的基础。本研究以大丽轮枝菌侵染后的棉花根系为材料,诱导抗病相关基因的表达,从而保证构建的文库基因信息更全面。RNA 质量是构建高质量cDNA 文库的前提,本试验中提取的28S 和18S rRNA 条带清晰,RNA 完整性较好,未发生降解,mRNA 也都被高效的反转录了。为提高文库质量,获得的双链cDNA 进行过柱纯化去除短片段,电泳检测条带基本均在0.5 kb以上,说明短片段已去除。通常以重组序列完整性和文库代表性来评价cDNA 文库质量[37],一般的cDNA文库库容须达到1×106CFU,文库滴度须达到1×108CFU.mL-1[29]。本研究构建的cDNA 文库库容大于2×106CFU,文库滴度大于2×109CFU.mL-1,重组率达到了94%,PCR 鉴定文库插入片段大小不一,多样性良好,但有个别文库克隆片段偏小,所能提供的基因信息少,可能需要几个克隆才能覆盖一个完整的全基因cDNA。总体来说,本试验中文库构建较成功,为后续筛选病原菌中和棉花互作蛋白奠定了基础。
黄萎病菌基因组编码700 多种假定调节宿主免疫的分泌蛋白[38]。VdSCP7 是一种新鉴定的大丽轮枝菌分泌蛋白,它定位于植物细胞核,且VdSCP7 诱导的植物免疫反应依赖于其在植物的核定位[39]。植物在真菌侵染过程中可以识别VdSCP7 并激活ETI 反应,增强抗病性,但目前尚不清楚这一过程的分子机制[27]。关于大丽轮枝菌一些效应因子如大丽轮枝菌蛋白激发子(PevD1) 的研究已多见报道[40-42],但棉花中与VdSCP7 互作蛋白的研究尚鲜见报道,筛选棉花中与VdSCP7 互作的蛋白,将有助于阐明这一作用机制。本研究构建了无自激活活性及毒性的诱饵载体pGBKT7-SCP7 可直接用于下一步酵母双杂交cDNA文库的筛选。
4 结论
本研究成功构建了高质量的棉花酵母双杂交cDNA 文库和无自激活活性及毒性的诱饵载体pGBKT7-SCP7,为进一步研究VdSCP7 作用机理奠定了基础。利用本研究构建的棉花cDNA 文库和酵母双杂交技术,可以大规模、快速筛选棉花抗病相关基因,为棉花和黄萎病菌互作机制的研究提供资源。
猜你喜欢
作物学报(2022年9期)2022-07-18
科普童话·神秘大侦探(2022年4期)2022-05-26
农业科技与信息(2021年4期)2021-12-05
新农村(浙江)(2021年2期)2021-11-30
猪业科学(2021年3期)2021-05-21
中华诗词(2019年1期)2019-11-14
阅读与作文(小学低年级版)(2017年10期)2017-10-27
农家科技下旬刊(2016年6期)2016-05-14
公务员文萃(2015年9期)2015-09-29
环球人物(2015年12期)2015-09-10
