免疫检查点抑制剂与抗血管生成药物合用治疗肿瘤的研究进展
2021-02-07 06:28:58郑维维杨春媚王爱云吴媛媛
中国药理学通报 2021年2期
郑维维,钱 程,邹 伟,杨春媚,张 珊,王爱云,2,陆 茵,2,吴媛媛,2
(南京中医药大学 1.药学院,江苏省中药药效与安全性评价重点实验室,2. 江苏省中医药防治肿瘤协同创新中心,江苏 南京 210023)
免疫检查点主要包括细胞程序性死亡受体-1(programmed cell death receptor 1,PD-1)和细胞毒性T淋巴细胞抗原-4(cytotoxic lymphocyte antigen 4,CTLA-4)。免疫检查点作为免疫稳态的重要组成部分,可下调机体免疫反应应答并参与外周耐受,而上调免疫检查点信号通路可使得肿瘤细胞免受免疫细胞监视[1]。因而免疫检查点受体及其配体是理想的抗肿瘤治疗靶点。但有研究发现通过阻断PD-1治疗肝癌患者,缓解率约为15%-20%[2-3]。
由于肿瘤细胞快速分裂增殖,血液供应与活性代谢无法满足要求,导致肿瘤微环境缺氧和酸中毒。低氧诱导肿瘤细胞和基质细胞分泌促血管生成因子,从而打破了促血管生成因子和抗血管生成因子之间的平衡,血管生成途径被激活,导致血管不成熟,且呈现迂回、渗漏的状态,肿瘤灌注不良。但抗血管生成药物会过度削减血管导致肿瘤组织缺氧加重,其中涉及到诸多相关蛋白或信号通路,而单一靶标的药物干预其靶点,往往会造成其他血管生成因子代偿性现象,从而恢复肿瘤血管新生,这些都是导致抗血管生成药物疗效不佳的原因[4]。Yasuda等[5]发现ICI和抗血管生成药物在治疗小鼠结肠癌具有协同作用。随后Wu等[6]证实ICI联合抗血管生成药物可以有效地延长乳腺癌小鼠的总生存期(overall survival,OS)。
本文对抗血管生成疗法作用机制、肿瘤血管生成对ICI治疗的影响以及ICI联合抗血管生成药物研究进展进行综述。希望通过本文能够加深对ICI联合抗血管生成药物治疗肿瘤的认识,并为后期的研究提供参考。
1 抗血管生成疗法
实体瘤会分泌多种促血管生成因子,例如血管内皮生长因子(vascular endothelial growth factor,VEGF)、血小板衍生生长因子(platelet-derived growth factor,PDGF)、肝细胞生长因子(hepatocyte growth factor,HGF)以及apelin等等,其中VEGF介导内皮细胞过度增殖使其在血管生成中发挥着核心作用。VEGF主要与其受体VEGFR2 结合,发挥生物学功能。一方面,VEGF-VEGFR2通过激活下游PLCγ-PKC-Raf-MAPK和Grb2-Gab1-MAPK / PI3K-Akt信号通路来促进血管性血友病因子(von Wilebrand Factor,vWF)的表达和内皮细胞的过度增殖和迁移[7];另一方面,VEGF-VEGFR2可以通过激活VEGFR2-TSAD-SRC-cadherin和PI3K-AKT-eNOS-NO信号通路导致血管渗漏[8]。
1.1 抗血管生成:从抑制血管生成到肿瘤血管正常化正常生理条件下,功能性血管的形成需要经历内皮细胞之间紧密连接,基底膜形成和周细胞包被新生血管等过程[9]。但肿瘤微环境中VEGF的持续分泌,导致肿瘤血管结构异常。Juda Folkman教授于1971年率先提出肿瘤生长和转移都依赖于肿瘤的血管,认为抑制肿瘤血管生成能够作为抑制肿瘤生长和延长带瘤生存期的治疗法则,但抗血管生成药物后期会导致肿瘤细胞转化为缺氧可耐受表型,导致它的侵袭性和转移能力增加[10]。
抗血管生成药物联合其他疗法显著抑制肿瘤生长[11]。但使用抗血管生成药物的同时,将会抑制药物和氧气的运输。由此,Jain[12]建立了一个模型来描述抗血管生成药物治疗时肿瘤血管的瞬时状态:血管正常化。在该模型中,当抗血管生成因子与促血管生成因子达到一定的平衡,异常结构和功能的肿瘤血管会逐渐转变为正常血管的表型。Huang等[11]通过研究抗血管生成剂量与疗效之间的关系,发现使用高剂量(120或150 mg·kg-1)的VEGFR2抑制剂虽然能显著抑制肿瘤血管生成,但会加速恶性肿瘤的转移,而在使用低剂量(10、20 mg·kg-1)或常规剂量(40 mg·kg-1)时,则具有抑制血管数量的作用,较低剂量的抗血管生成药物在诱导肿瘤血管正常化方面优于较高剂量的疗效,血管正常化状态取决于给药时间和剂量。
此外,apelin是血管紧张素受体样蛋白J(angotensin protein J,APJ)受体的内源性配体,作为一种促血管生成肽,在多种肿瘤细胞以及肿瘤内皮细胞中APLN/APLNR途径均被上调,且其与肿瘤治疗的不良预后相关[13]。Iris等[14]发现在肺癌和乳腺癌模型中,敲除apelin可以弥补抗血管生成疗法因缺氧带来的耐药相关性问题。
1.2 血管正常化改善肿瘤免疫微环境肿瘤血管正常化对于肿瘤微环境具有调节作用,能够将免疫抑制状态重编程为免疫支持状态。异常的肿瘤血管新生上调髓源抑制细胞(myeloid-derived suppressor cells,MDSC)、树突状细胞(dendritic cells,DC)还有肿瘤细胞的PD-L1的表达水平,抑制效应T细胞对肿瘤的浸润并促进肿瘤相关巨噬细胞(tumor-associated macrophage,TAM)极化为免疫抑制性M2样表型,形成低氧、酸性的肿瘤微环境。此外,缺氧还可以上调免疫抑制生长因子,例如转化生长因子(transforming growth factor-β,TGFβ)。当给药时间和剂量都适宜时,抗血管生成疗法能够促进肿瘤血管正常化,增加效应T细胞的浸润、减少Treg细胞以及MDSC募集,缓解的缺氧优先诱导TAM极化为M1样表型[15]。此外,低氧诱导的免疫抑制性信号如PD-L1通过血液灌注的改善也得以下调[16]。另外,抗VEGF药物可阻断DC分化的抑制信号并降低肿瘤微环境中MDSC数量[17]。
2 肿瘤血管生成对ICI治疗的影响
2.1 肿瘤浸润淋巴细胞的状态决定ICI疗效对于ICI治疗,尤其是抗PD-1 / PD-L1抑制剂干预,肿瘤微环境中存在肿瘤浸润淋巴细胞(tumor infiltrating lymphocyte,TIL)是有效治愈肿瘤的必要条件。根据已存在的TIL状态,将肿瘤微环境分为3种类型:①免疫炎症类型,功能性CD8+T细胞密集浸润;②无浸润类型,异常的血管生成和免疫抑制微环境阻止T细胞的浸润;③免疫低下类型[18]。已经证实,属于免疫炎症型的肿瘤比其他两种类型的肿瘤对ICI治疗更敏感[19]。因此,提高肿瘤组织T细胞浸润程度能够增强ICI疗效。
2.2 血管生成促进免疫抑制微环境的形成在肿瘤免疫过程中,抗原呈递决定特异性T淋巴细胞的生成。有特定T细胞受体(T cell receptor,TCR)的T细胞浸润到肿瘤,才能进行抗原识别并杀伤肿瘤。实体瘤内的血管生成亢进通过多种途径影响免疫抑制微环境的形成。VEGF和血管生成素-2(angiopoietin-2,Ang-2),可直接或间接作用内皮细胞以及先天性和适应性免疫细胞发挥免疫抑制作用。一方面,VEGF和Ang-2增强内皮细胞中PD-1和PD-L1的表达,导致表达PD-1的细胞毒性T细胞功能丧失。其次,VEGF下调肿瘤血管细胞粘附分子-1(vascular cellular adhesion molecule-1,VCAM-1)的表达,进而抑制T细胞穿越内皮层渗入肿瘤。而且,由于Treg细胞中的FLICE抑制蛋白(cellular FLICE/caspase-8 inhibitory protein,CFLIP)高表达,肿瘤内皮细胞(endothelial cells,EC)Fas配体能够选择性触发表达Fas的CD8+T细胞凋亡[15]。最后,EC还通过上调CLEVER-1 / stabilin-1募集免疫抑制性Treg细胞。所有这些途径诱导有效屏障的形成,导致细胞毒性T细胞不能浸润肿瘤[15]。另一方面,VEGF通过免疫细胞中的VEGFR2信号进行传导。首先,它可以上调树突状细胞PD-L1中的表达,并由于其抗原呈递能力受损而抑制树突状细胞的成熟和功能。其次,VEGF / VEGFR2信号传导抑制效应T细胞的增殖,增加并促进Treg的肿瘤归巢,这种作用取决于neuropilin-1(nrp-1)。而且除了PD-1、CTLA-4外,VEGF还可以上调T细胞其它负性免疫检查点受体,导致T细胞衰竭。最后,高水平VEGF、Ang-2以及其他肿瘤分泌因子会促进TAM、MDSC和未成熟树突状细胞的募集和增殖。此外,这些免疫细胞群所分泌的细胞因子会促进肿瘤血管生成和免疫抑制微环境的形成[15]。
2.3 免疫细胞调节血管生成一方面,免疫抑制细胞群如Treg、MDSC和M2型TAM,有助于肿瘤免疫逃逸,通过分泌基质细胞衍生因子1和胎盘生长因子(placental growth factor,PIGF)促进肿瘤血管生成。另一方面,TAM和MDSC通过表达基质金属蛋白酶(matrix metalloproteinases,MMP),以降解细胞外基质去诱导肿瘤血管生成。Rolny等[20]发现富组氨酸糖蛋白(histidine-rich glycoprotein,HRG)通过抑制巨噬细胞FcγR受体,下调其衍生的PIGF,诱导M2样TAM极化为M1样表型,从而促进肿瘤血管正常化和减缓肿瘤的生长与转移。此外,活化的CD11b+Gr1loF4/80+Siglec-F+嗜酸性粒细胞促进肿瘤血管正常化并有助于CD8+T细胞对肿瘤的杀伤作用。但其确切机制尚不明确,可能是通过嗜酸性粒细胞来源的干扰素-γ(interferon-γ,IFN-γ)和肿瘤坏死因子(tumor necrosis factor,TNF)使得TAM极化为M1样表型,导致VEGF的表达降低[21]。
此外,Huang等[22]发现单独使用ICI通过激活效应T细胞达到促进肿瘤血管正常化的效果[22]。肿瘤血管正常化能减轻肿瘤微环境免疫抑制的过程,促进效应T细胞浸润。免疫微环境重编程与肿瘤血管正常化之间的这种正反馈调节相互加强,促进免疫介导的肿瘤根除[22]。
3 ICI联合抗血管生成药物的研究进展
活化的T细胞通过分泌IFN-γ作用于肿瘤血管内皮细胞上的IFN-γ受体上,直接促进使肿瘤血管正常化[22]。基于肿瘤免疫与血管生成之间的相互作用,推测抗血管生成药物可能增强ICI的疗效。除了降低肿瘤微环境间质压并改善T细胞浸润外,我们发现联合治疗也与其它机制相关。
3.1 阻断VEGFR2诱导的免疫检查点表达Dan等[23]进行肝癌小鼠模型研究。构建原位移植或诱导HCC模型,分为6组(Ⅰ)IgG;(Ⅱ)抗PD-1 单克隆抗体(monoclonal antibody,mAb):RMP-014;(Ⅲ)抗VEGFR2 mAb:DC101 10 mg·kg-1;(Ⅳ)抗VEGFR2 mAb:DC101 40 mg·kg-1;(Ⅴ)抗PD-1 mAb +抗VEGFR2 mAb:DC101 10 mg·kg-1;(Ⅵ)抗PD-1 mAb +抗VEGFR2 mAb:DC101 40 mg·kg-1。其中联合治疗组具有最佳疗效。为了证实VEGFR2对免疫检查点表达的影响,采用体外3-D培养,构建异型肿瘤类器官。使用DC101后,类器官中肿瘤细胞和基质细胞中PD-L1表达明显上调。随后磁珠分选VEGFR2+细胞探究VEGFR2阻断后PD-L1上调的具体机制,表明DC101靶向内皮细胞中VEGFR-2上调HCC细胞PD-L1这一过程,部分由内皮细胞表达的IFN-γ所介导[23]。
此外,Voron等[24]观察到抗VEGF可以选择性抑制肿瘤内CD8+T细胞免疫检查点的表达。VEGF可以通过激活VEGFR2-PLCγ-calcineurin-NFAT信号通路来上调PD-1的表达。因此联合疗法能有效阻断PD-1/PD-L1轴,抑制肿瘤生长,特别是VEGF分泌较高的肿瘤。
3.2 诱导高内皮小静脉形成Allen等[25]研究抗PD-L1 mAb:B20S和抗VEGFR2 mAb:DC101在胰腺神经内分泌肿瘤、乳腺癌和胶质母细胞瘤的小鼠模型中联合治疗的疗效。在胰腺神经内分泌肿瘤和乳癌中,联合疗法在肿瘤体积和总生存期方面比单一疗法显示出优势,对于胶质母细胞瘤而言,联合疗法优势更显著。联合治疗2周后,仅50%胶质母细胞瘤中IFN-γ+CD8+T细胞略微增加,而胰腺神经内分泌肿瘤和乳腺癌中IFN-γ+CD8+和IFN-γ+CD4+T细胞水平增加了两倍。胰腺神经内分泌肿瘤和乳腺癌中的血管除了完整的周细胞覆盖之外,由肥大的内皮细胞而不是扁平的内皮细胞增厚,表现出高内皮小静脉(high endothelial venules,HEV)特征[26]。HEV与淋巴细胞归巢有关,淋巴毒素β受体(lymphotoxin beta receptor,LTβR)信号通路对维持HEV表型至关重要,联合治疗通过激活LTβR信号通路有效杀死胶质母细胞瘤细胞。
3.3 抑制IFN-γ介导的负反馈调节Schmittnaegel等[27]发现双特异性抗体A2V同时靶向VEGF和ANG2的阻断要比单一疗法有更好的治疗效果,且联合抗PD-1治疗能进一步增强双重阻断的疗效。在这项临床前研究中,采用多种荷瘤小鼠模型,包括转基因或移植性乳腺癌、胰腺神经内分泌癌、黑色素瘤和结直肠癌模型。A2V处理后,肿瘤微环境中多种抗肿瘤免疫细胞数量增加,如成熟DC、M1样表型TAM、IFN-γ+/ CD69+CD8+T细胞等,但由于IFN-γ所介导的负反馈调节机制,致使肿瘤血管周围的CD8+T细胞增加且肿瘤细胞PD-L1的高表达。而抗PD-1和A2V的联合治疗能够抑制IFN-γ介导的负反馈调节并增强免疫反应,与A2V疗法相比,接受联合治疗的超过30%的小鼠OS延长。
3.4 ICI联合抗血管生成药物的临床研究
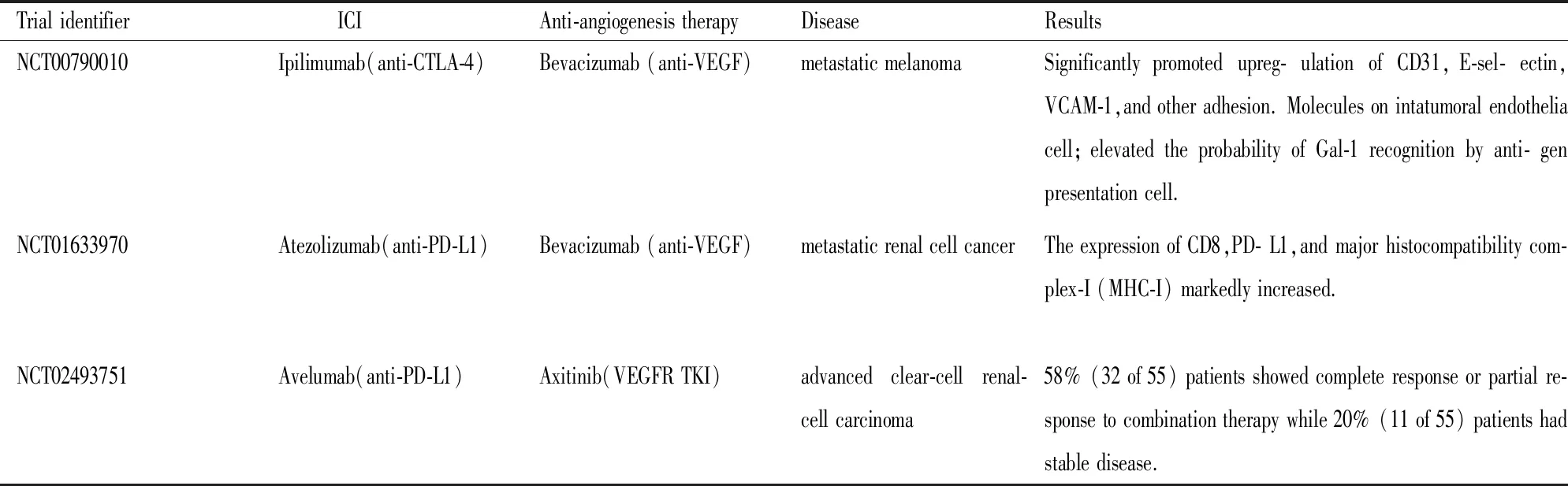
3.4.1抗CTLA -4 mAb联合抗VEGF mAb NCT00790010是一项I期临床试验[28],旨在探讨易普利姆玛(抗CTLA-4 mAb)和贝伐单抗(抗VEGF mAb)在转移性黑色素瘤患者中的作用。所有46名入选患者均被分为4组,并接受了不同剂量的联合治疗。该治疗方案显著上调肿瘤血管内皮细胞的CD31、E-选择素、VCAM-1和其他粘附分子的表达,增加细胞毒性T细胞和成熟DC浸润。与之前的研究结果相比,接受联合治疗的患者在预后方面占有很大优势。
进一步的研究表明,联合治疗的有利优势可能源于诱导galectin-1(Gal-1)的免疫反应[28]。Gal-1参与肿瘤细胞增殖、侵袭、免疫逃逸和血管生成等过程。收集患者的血浆样品以检测抗Gal-1抗体的浓度,结果表明,联合治疗后62.5%完全缓解/部分缓解患者中抗Gal-1抗体滴度增加≥1.5倍,36.4%病情稳定患者和23.1%病情恶化患者治疗后抗Gal-1抗体滴度略微增加。对联合疗法的不同反应归因于不同的抗Gal-1免疫反应。一方面,抗VEGF mAb上调Gal-1的表达,另一方面,抗CTLA-4 mAb增加T细胞克隆的表型。这两个因素提高了抗原呈递细胞识别Gal-1的可能性。此外,另外两项临床试验(NCT02210117和NCT01950390)正在研究易普利姆玛联合贝伐单抗分别治疗转移性肾癌和III-IV期黑色素瘤患者的疗效。
3.4.2抗PD- L1 mAb联合抗VEGF mAb Wallin等[29]受到抗CTLA-4 mAb和抗VEGF mAb联合治疗作用显著的启发,开展抗PD-L1 mAb联合抗VEGF mAb的临床研究。NCT01633970是一项旨在研究阿特珠单抗(抗PD-L1)联合贝伐单抗或化疗的安全性和药理学作用的1b期临床实验。
10名肾细胞癌转移性患者接受1周期的贝伐单抗单药治疗后,实施联合治疗方案,直至疾病向不良方向发展。结果10名患者中有8名患者病情得以控制。此外,与贝伐单抗单药治疗后患者的肿瘤样品相比,联合治疗后CD8、PD-L1以及主要组织相容性复合物-I的表达显著增加[29]。
3.4.3抗PD-L1 mAb联合抗血管生成药物TKI 到目前为止,临床研究中的联合方案多由ICI和抗血管生成药物贝伐单抗组成。2018年,Choueiri等[30]首先报道了阿维鲁单抗(抗PD-L1 mAb)联合阿昔替尼(TKI)在晚期肾透明细胞癌(JAVELIN Renal 100)中的临床实验(NCT02493751)。55名患者中,除一名患者由于磷酸肌酸激酶异常增加,其余54名患者都接受阿维鲁单抗联合阿昔替尼治疗。在近一年的随访期内,58%的患者对联合治疗表现出完全缓解或部分缓解,20%的患者病情相对稳定。此外,发现PD-L1表达没有显著影响治疗效果[30],见Tab 1。
Tab 1 Clinical trials investigating efficacy of ICI plus anti-angiogenesis therapy
4 总结与展望
免疫检查点抑制剂和抗血管生成药物单独使用均有各自的局限性,而临床前和临床研究表明抗血管生成药物联合ICI治疗肿瘤的作用相互增强。一方面,抗血管生成剂通过增加抗/促肿瘤免疫细胞的比例和降低多个免疫检查点的表达抑制负性免疫应答;另一方面,ICI治疗可以重塑免疫微环境并促进血管正常化。此外,血管正常化能够提高药物递送效率,使得较小剂量的ICI就能发挥较大治疗效果,避免大剂量ICI带来的不良反应。但目前需要解决的主要问题是如何在联合治疗中优化抗血管生成剂的给药时间和剂量,扩大血管正常化窗口并获得最长的生存期。总之,ICI联合抗血管生成药物将是克服治疗耐药性并改善患者预后的有效策略。
猜你喜欢
日本研究(2023年2期)2023-11-29 12:16:44
计算机系统应用(2022年4期)2022-05-10 08:41:10
天津医科大学学报(2021年4期)2021-08-21 02:14:52
现代畜牧科技(2021年7期)2021-07-28 06:41:00
国际呼吸杂志(2019年4期)2019-03-12 01:08:18
兽医导刊(2019年1期)2019-02-21 01:13:54
人民中国(日文版)(2016年10期)2016-08-23 11:21:14
中国药理学与毒理学杂志(2015年3期)2015-12-16 09:11:40
齐鲁周刊(2015年38期)2015-12-11 09:23:52
现代计算机(2015年31期)2015-09-28 05:31:51