
Bart综合征一例基因检测
2021-02-06 12:06刘静朱静
中国麻风皮肤病杂志 2021年2期
刘 静 朱 静
1淮北市人民医院(徐州医科大学淮北临床学院)皮肤科,安徽淮北,235000;2淮北市人民医院(徐州医科大学淮北临床学院)烧伤整形科,安徽淮北,235000
临床资料患儿,男,于2020年2月7日经我院产科顺产娩出。胎龄(40周+1),出生体重4.1 kg,产后即发现患儿双下肢、足部、左手腕、面部皮肤缺失,于2月8日转上级医院进一步治疗。入院查体:一般情况可,各系统未见异常。面部部分皮肤缺失伴少许渗出,口腔黏膜破溃,双下肢及足部伸侧皮肤大面积缺损、糜烂、渗出,右侧足背过伸(图1)。
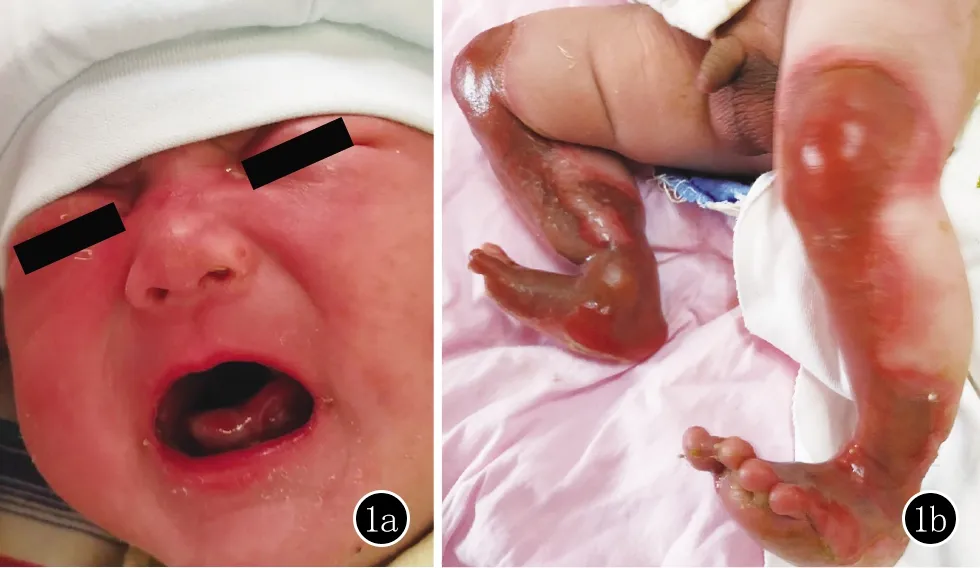
图1 1a:口腔黏膜受损;1b:双下肢皮肤破损伴少量渗出,右下肢过伸
辅助检查:2020年2月10日血常规示白细胞计数4.44×109/L,中性粒细胞百分比35.00%,红细胞计数6.08×1012/L,血红蛋白218 g/L;血生化:总胆红素48.00 μmol/L,直接胆红素16.00 μmol/L,间接胆红素32.00 μmol/L,白蛋白33.2 g/L,C-反应蛋白69.80 mg/L;脓毒血症二项(PCT/IL-6):降钙素原0.630 ng/mL,白介素6181.50 pg/mL;免疫十一项:乙肝表面抗体阳性。2020年2月12日血常规:白细胞计数5.94×109/L,中性粒细胞百分比31.00%,红细胞计数5.71×1012/L,血红蛋白204 g/L;血生化:C-反应蛋白59.90 mg/L;脓毒血症二项(PCT/IL-6):降钙素原0.164 ng/mL,白介素6338.80 pg/mL。2020年2月11、12、14日在院期间先后3次取创面分泌物送检普通细菌培养2天,均无细菌生长。
经询问患儿父母双方,均否认有相关家族史,家族成员在出生时无皮肤异常、指趾甲异常,非近亲结婚。患儿为头胎顺产,既往无流产史。孕妇在孕期按期孕检,孕中期行彩超检查时提示:胎儿双侧肾窦分离,胎儿双侧足背过度前屈,NT、无创、OGTT等均未见明显异常。
患儿家长拒绝皮肤组织病理有创性检查。根据患儿的临床表型可初步诊断其为大疱性表皮松解症。为进一步明确诊断,经患儿父母同意,对患儿进行基因检测。采集患儿2 mL外周血提取基因组DNA,通过二代测序对大疱性表皮松解症已知的21个致病基因(KRT5、KRT14、PLEC、DST、KLHL24、TGM5、DSP、PKP1、JUP、EXPH5、COL7A1、LAMA3、LAMB3、LAMC2、ITGA6、ITGB4、COL17A1、CD151、ITGA3、PLCG2、FERMT1)的全部外显子和外显子侧翼序列进行检测。结果在先证者(患儿)COL7A1基因上检测到2个致病性变异,即c.2005C>T:p.R669X和c.7922G>A:p.G2641E。而在另外20个基因上未检测到致病性变异。进一步进行一代测序验证,显示先证者这2个变异分别来自其母亲(c.2005C>T)和父亲(c.7922G>A)(图2)。COL7A1基因是常染色体大疱性表皮松解症的明确致病基因,因此,根据患儿的临床表型和基因检测结果,可明确诊断隐性营养不良型大疱性表皮松解症(recessive dystrophic epidermolysis bullosa, RDEB),即Bart综合征(Bart’s syndrome, BS)。

图2 一代测序验证:先证者检测到c.2005C>T和c.7922G>A突变;父亲检测到c.7922G>A突变;母亲检测到c.2005C>T突变
治疗:患儿经换药等支持对症处理于一周后出院,双下肢部分伸侧面及其他区域的表皮缺损外渗出量减少,见新鲜表皮被覆(图3)。

图3 患儿出院后双下肢及足部表皮缺损处渗出减少,见有新鲜表皮被覆
讨论先天性皮肤缺损又名皮肤再生不良(aplasia cutis congenita, ACC)。先天性皮肤缺损呈现多部位发病,约有84%位于头部[1],70%是单发皮损,20%为多发或伴有其他部位的皮损。缺损形状呈块状、片状或不规则形状[2]。先天性大疱性表皮松解症指患儿身体的某处表皮、真皮甚至皮下组织出现先天性缺损或发育不全,皮肤受压或摩擦后即可引起大疱,多发生于四肢末端和关节伸侧的机械损伤后出现水疱、大疱、愈后瘢痕、粟丘疹和指(趾)甲损害是共同特征。严重者水疱不仅局限于体表皮肤,而且可见于体内的软组织(黏膜)[3]。注意到本例患儿的皮损集中分布在双下肢、手腕部,且有口腔黏膜受累,与国外文献多报道皮损多发在头部存在差异,可以解释为区域差异导致[4]。
1986年Frieden[5]根据皮损部位、临床表现、是否合并其他异常将ACC分为9类。其中第6类:皮损位于四肢,伴有大疱性表皮松解症,水疱多位于四肢,即ACC同时伴随大疱性表皮松解症(epidermolysis bullosa,EB)。
大疱性表皮松解症是一种分型复杂的疾病,亚型较多,而且几种亚型有时同时出现于同一患者,即使是同一患者也可能在不同时期表现为几种亚型特征,因此多数临床医生难以做出准确诊断[6]。2013年国际EB会议审核了本病的整体数据,强调了不再仅仅依靠临床特征进行亚型分类,即修改了EB患者的分型方法,更多地关注每种亚型在分子层面的发病原因,倡导在临床上使用新的分型和诊断方法:剥洋葱法[7]。这种方法采用渐进的方式,先根据靶蛋白和超微结构中不同皮肤裂隙位置划分到四种主要类型开始,即分为表皮内是单纯型EBS(EB simplex)、基底膜中是交界型JEB(junctional EB)、基底膜下是营养不良型DEB(dystrophic EB)和混合模式Kindler综合征(KS)4个型别,且每个型别又有多种亚型。四种主要类型的区别:单纯型EBS的裂隙在基底层角化细胞区,水疱位于表皮的中层或上层;交界型JEB的裂隙发生在透明板内;营养不良型DEB水疱发生在致密板下面的真皮上部;Kindler综合征(KS)的裂隙可能发生在基底层角化细胞中,透明板层或致密板下面[8]。早期诊断多以透射电镜确认患者水疱所在位置,然后再进一步明确诊断;近年逐渐转变为使用免疫荧光或免疫组化定位[9],做相应基因测序以确定致病基因位点,对患者的 EB 类型、遗传模式、临床表现、免疫荧光抗原定位检测结果及突变情况等因素进行综合考虑。在可能致病基因位点上再进行基因定位诊断,最后确认EB分型[10]。
研究表明,EB的发病原因是角化细胞或皮肤基底膜区(表皮和真皮的交界)内的蛋白质突变引起皮肤超微结构异常,通常皮肤和皮外症状的严重程度反映了突变的类型和目标蛋白的超微结构位置。所有的营养不良型DEB(dystrophic EB)的突变都发生在VII型胶原基因[11]。
Bart综合征是营养不良型大疱性表皮松解症的一种,非常罕见,发病率大约在3.3/106,1966年由Bart首先报道[12]。1996年Chrlstiano等证实本病是VII型胶原基因(COL7A1)突变所致[13],目前已发现COL7A1基因的700多个突变。本例中在患儿COL7A1 上检测到的c.2005C>T和c.7922G>A复合杂合变异,前者为无义突变,会导致肽链延伸至第669位氨基酸位置时提前终止,在ExAC(EAS)中的频率为0.0001,在NCBI和ClinVar数据库中均记录为Pathogenic(rs780261665);后者为错义突变,会导致肽链第2641位氨基酸由甘氨酸转变为谷氨酸,在ExAC(EAS)中的频率为0,在NCBI、Ensembl、ClinVar等数据库中未见报道。基因突变导致pro-α1(VII)链蛋白的生产出现异常,从而影响VII型胶原蛋白的生产,继而影响锚纤维的组成和功能[14],最终造成皮肤非常脆弱,摩擦或其他轻微外伤可能导致两层皮肤分离,易起水疱。
Bart综合征的临床诊断需与其他疾病鉴别,如葡萄球菌性烫伤样皮肤综合征、新生儿脓疱疮、先天性大疱性鱼鳞病样红皮病、先天性梅毒、新生儿单纯疱疹等。其中葡萄球菌皮肤烫伤样综合征(staphylococcal scalded skin syndrome, SSSS)是发生于新生儿的严重急性泛发性剥脱性脓疱病[15],是在全身泛发红斑的基础上,发生松弛性烫伤样大疱及大片表皮剥脱为特征。多发生于出生后1~5周的婴儿,发病突然,初在口周或眼睑四周发生红斑,其后迅速蔓延至躯干和四肢近端,甚至泛发全身,一般经过7~14天后痊愈。本例患儿除特异性皮损外,并无明显的感染性中毒表现,故而可排除以上感染性疾病的可能性。
大疱性表皮松解症的预后情况高度依赖于其疾病亚型,差异较大。其中单纯型EBS(EB simplex)和显性营养不良型(Dominant dystrophic epidermolysis bullosa,DDEB)预后较好,可存在正常的预期寿命。交界型JEB(junctional EB)因水疱全身泛发、广泛剥脱,预后通常不好,可在出生后早期即死亡[16]。Bart综合征通常出现手指和脚趾均被瘢痕包裹,随年龄增长肌肉逐渐萎缩,最终形成棒状手而致残,同时可伴黏膜损害、甲营养不良、牙齿排列不规则,毛发稀疏等。严重病例因为大面积表皮剥脱、黏膜受累、伴发心肌病而死亡。少数RDEB患儿可出现恶性黑素瘤[17]。因此对EB的分型诊断应尽量、及早确定,以利于后期针对性治疗。
目前对该病治疗尚无有效方法,多采取提前干预,对可能发生严重的皮肤及口腔感染,局部渗出液增多引起的低蛋白血症、电解质紊乱、肺部感染等制订出护理计划,并严格按照执行,纠正营养不良,防止细菌感染,保护表皮免受摩擦,减少身体残疾和肌肉挛缩,以预防并发症的发生,促进皮损早期愈合。本例在治疗上予以头孢美唑抗感染,患儿体表皮肤缺损面实施无菌换药,使用优拓,系脂质水胶敷料,由100%聚酯网、水胶体(羧甲基纤维素钠)、凡士林构成,创造出有益于伤口愈合的湿性环境,并及时祛除死皮和脓痂,再涂以抗生素类及重组人表皮生长因子凝胶,凡士林覆盖,采取每日或隔日换药一次。创面处保持干燥,尽可能避免长时间受压,同时积极预防继发细菌感染。
因BS目前仍缺乏有效治疗手段,即使经过长期治疗,依然严重影响患者的生活质量,则更加强调了分子生物学、遗传学研究的重要性,因此产前诊断是有效的预防措施,除常规孕检外,应积极开展针对性的遗传咨询和指导。预先尽可能明确父母或其他已患病亲属的致病性基因突变点,在妊娠第9~10周做羊膜腔穿刺,获取绒毛膜绒毛或羊水细胞标本进行DNA产前检测,从而促进优生优育。
志谢:感谢上海安百隆生物科技有限公司技术部孔祥生老师在基因检测方面所给予的指导及帮助!
猜你喜欢
临床骨科杂志(2022年3期)2022-06-24
今日农业(2022年3期)2022-06-05
中国生物制品学杂志(2022年3期)2022-05-13
建材发展导向(2021年14期)2021-08-23
皮肤病与性病(2021年3期)2021-07-30
爱你·健康读本(2019年8期)2019-11-22
爱你(2019年29期)2019-11-07
祝您健康·文摘版(2019年4期)2019-06-11
环球市场信息导报(2018年21期)2018-07-27
