
I掺杂TiO2纳米管阵列平面光催化燃料电池的性能研究
2021-02-06 13:39:54席清华黄宜强
华东师范大学学报(自然科学版) 2021年1期
周 君,席清华,黄宜强,聂 耳,孙 卓
(华东师范大学 纳光电集成与先进装备教育部工程研究中心, 上海 200062)
0 引 言
工农业的迅速发展产生大量污水, 排入自然界引发了严重的水体污染, 对人类的生产生活造成了严重威胁, 废水处理成为全世界共同关注的问题[1]. 对于水体污染, 常用的处理方法有活性污泥法、物理吸附法、催化氧化法等[2], 其中, TiO2由于其优异的光催化活性, 被广泛地应用在光降解领域[3], 但是其禁带宽度较宽, 仅能对紫外光进行响应, 光生电子–空穴(e–-h+)对复合率较高, 因此光催化降解性能较低[4].
光催化燃料电池(Photocatalytic Fuel Cell, PFC)集成了光催化和燃料电池的优点, 具备了光催化污染降解和能量回收的特点, 是同时解决废水污染问题和能源短缺问题的理想方案之一[5-6], 在降低光生电子–空穴复合率方面显示出巨大的优势[7-8]. PFC系统主要由光阳极、光阴极、电解质和外部电路组成[9]. 光阳极使用半导体材料作为催化剂, 在受到一定波长的光辐射条件下, 产生e–-h+对. h+可以将OH–氧化为具有强氧化性的·OH, 而光生电子由外电路流向阴极, 因此可以有效防止光生电子-空穴对的复合[10-12].
过去十几年科研工作者投入大量精力研发阳极材料, 这些工作的主要目的是提高可见光利用率和光氧化速率. 其中, 卤素掺杂能有效提高光催化剂的光学和表面性能, 常见的卤素掺杂比如F、Cl、Br、I掺杂TiO2[13]. 文献报道I掺杂TiO2的可见光吸收能力要高于F掺杂TiO2[14-15], 这是由于I5+的离子半径与Ti4+非常接近, 易于取代TiO2中的Ti4+[16]. 在掺杂过程中, I5+会发生歧化反应生成I7+和I–[17],I5+/I7+能捕获光生电子, 从而降低光生电子–空穴的复合率[18]. 目前, 通常采用水热法制备I掺杂TiO2粉末, 并采用丝网印刷涂覆于FTO导电玻璃基体上[19], 由此制备的薄膜电极电阻大, 导电性较差.
此外, 光传输、底物传输及离子传输等过程在PFC中也起到重要的作用[20], 这些传输过程与电池的结构有着内在联系, 但是PFC的结构尚未得到广泛的探索. 传统的PFC需要用离子交换膜将其分为阳极和阴极两个腔室, 成本较高; 阴阳极距离较远, 传质受限, 内阻较大; 并且由于PFC体积过大,光还未到达催化剂表面就已有较大的衰减, 造成光子的吸收减少[21-24].
基于以上问题, 本文对PFC的结构进行了探索, 将光阳极与阴极平行放置, 得到平面型光催化燃料电池(planar Photocatalytic Fuel Cell, p-PFC), 并与传统的面对面光催化燃料电池(face-to-face Photocatalytic Fuel Cell, f-PFC)进行了对比. 采用阳极氧化法对钛片进行电解, 得到二氧化钛纳米管阵列电极(TNA), 并且在电解液中添加碘酸, 制备了I掺杂的钛纳米管电极(ITNA). 以Pt电极作为阴极, 选取了亚甲基蓝(Methylene Blue, MB)为降解物, Na2SO4溶液为电解液, 对影响p-PFC降解效率的影响因素进行了探究.
1 实验部分
1.1 TiO2光阳极的制备
光阳极的制备采用阳极氧化法, 将纯钛箔(国药集团, TA1级, 厚度0.1 mm)切割成2 cm × 10 cm的长方形钛片, 用180目砂纸打磨光滑后, 再用丙酮、乙醇及去离子水分别超声20 min, 于空气中自然干燥. 将盐酸与氢氟酸按体积比9∶1配置成浓溶液, 将钛箔浸没于酸溶液中清洗至表面光洁, 然后迅速置于去离子水中以洗净钛箔表面的酸残留, 最后置于空气中自然干燥.
将2 g氟化铵溶解于8 g去离子水中, 然后加入190 g乙二醇混合均匀, 得到电解液. 以钛箔为正极,铂电极为负极, 正负极间距为2 cm, 电压为30 V, 阳极氧化2 h, 取出用去离子水洗净钛箔表面的电解液, 自然干燥后置于马弗炉中, 以2 ℃·min–1的升温速率升温到500 ℃, 保温120 min, 得到TNA电极.将0.88 g碘酸溶解于电解液中, 其余步骤与制备TNA光阳极相同, 得到ITNA光阳极.
1.2 PFC组装
以制备的ITNA光阳极作为阳极, 铂电极为阴极, 将阴极与阳极固定于石英反应器中, 构筑PFC体系. 实验中光阳极面积为20 cm2, 光阴极面积为7 cm2, 根据阴阳极放置位置和光照方向设置了p-PFC和2种常见f-PFC结构, 如图1所示. 第一种f-PFC-1如图1a)所示, 纳米管一侧朝向阴极,光线从阴极方向垂直射向阳极; 第二种f-PFC-2如图1b)所示, 纳米管一侧朝向光源, 光线垂直射向光阳极. p-PFC结构如图1c)所示, 光阳极与阴极置于同一平面, 光线垂直射向光阳极. PFC装置外部用导线直接相连.
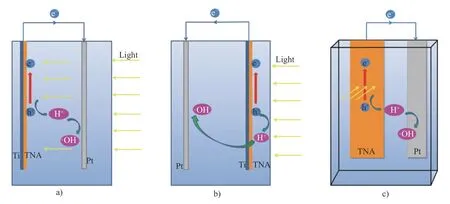
图1 不同电池结构示意图Fig. 1 Schematic diagram of different cell structures
1.3 光阳极表征
制备的样品采用场发射扫描电镜(FESEM, Gemini 450)分析微观形貌; 采用X射线电子衍射(XRD, Bruker D8 Advance, 铜靶Ka辐射)分析晶体结构; 采用能谱仪(EDS, Oxford6498)分析样品的元素分布; 采用X射线光电子能谱仪(XPS, Kratos Axis Ultra)分析样品的元素组成和价态结构.
1.4 光降解实验
在MB溶液中加入0.1 mol·L–1的Na2SO4作为电解质增强导电性能. 采用光催化反应仪器(上海比朗, BL-GHX-V, 光源为450 W金卤灯)进行光降解, 光源外设置滤光片, 仅保留可见光波段. 实验温度由冷却水循环系统控制在25 ℃左右. 首先避光60 min以达到吸附—解吸平衡, 之后打开光源,间隔一定时间取样记录, 用紫外分光光度计测定样品吸光度, 计算其降解效率.
2 结果与讨论
2.1 形貌与结构
图2为TNA 和ITNA电极的扫描电镜图和元素分布图. 从TNA极板的表面可以看到规则的孔洞结构, 内径为30 ~ 50 nm, 且分布较为均匀. 从ITNA的表面和局部放大图中可以看出, ITNA的表面结构无明显变化, 侧面是规则的纳米管管状结构, 有利于电极表面对有机污染物的吸附. 从ITNA的EDS图可以看出, Ti、O、I元素分布均匀, 没有明显的团聚现象, Ti、O、I的质量百分比分别为45.7%、50.99%、3.31%, 说明制备的电极杂质较少. I与Ti的原子百分比分别为0.63%和22.89%,得到I掺杂量为2.75%.
图3为纯钛片、TNA及TNA的X射线衍射图谱. 从图3中可以看到, 在25.3°、37.1°、37.9°、38.6°、48.1°、54.1°、55.2°及62.8°处存在明显的衍射峰, 分别对应锐钛矿相TiO2的(101)、(103)、(004)、(112)、(200)、(105)、(211)、(204)晶面(JCPDS 73-1 764)[25], 这说明制备的样品均有良好的结晶性. ITNA与TNA的衍射峰位置并没有发生偏移, 表明在ITNA表面没有I单质或I氧化物的存在. 没有其他晶型的TiO2衍射峰, 说明I掺杂不会影响TiO2的晶体结构. 钛片基底的XRD图谱仅包含Ti单质的吸收峰, 没有出现锐钛矿TiO2的吸收峰, 说明TNA与ITNA完全是由阳极氧化生成的.
为确定I是否掺杂到了TNA中, 采用XPS对ITNA进行了化学价态和表面元素组成的分析. 图4a)为ITNA的XPS总谱, 从图中可以看到Ti、O、I元素的特征峰, 而XRD中没有I的衍射峰, 说明I成功掺杂到TNA中. 图4b)为I 3d的高分辨图谱, 在620.5、621.7、623.2和624.9 eV处拟合出了4个特征峰, 分别对应I2(I-I键)、I–(I-H键)、I5+(I-O键)[16,26]和I7+[14]. I5+离子半径为0.062 nm, Ti4+离子半径为0.064 nm, 半径相差不大, 因此I5+容易取代Ti4+进入二氧化钛的晶格位点[27]. 从XRD图谱中可知ITNA的晶体结构没有发生变化, 而I–的离子半径为0.216 nm, O2–的离子半径为0.124 nm, I–的离子半径较大, 如果置换了Ti4+或O2–, 会改变TiO2的晶体结构, 因此I–可能位于晶体表面[16]. 图4c)为Ti 2p的高分辨图谱, 在458.0 eV、463.5 eV处存在两个特征峰, 分别对应Ti 2p3/2、Ti 2p1/2, 证实了Ti4+-O键的存在[28].

图2 TNA、ITNA 的SEM图和EDS元素分布图Fig. 2 SEM images and EDS mapping of TNA and ITNA
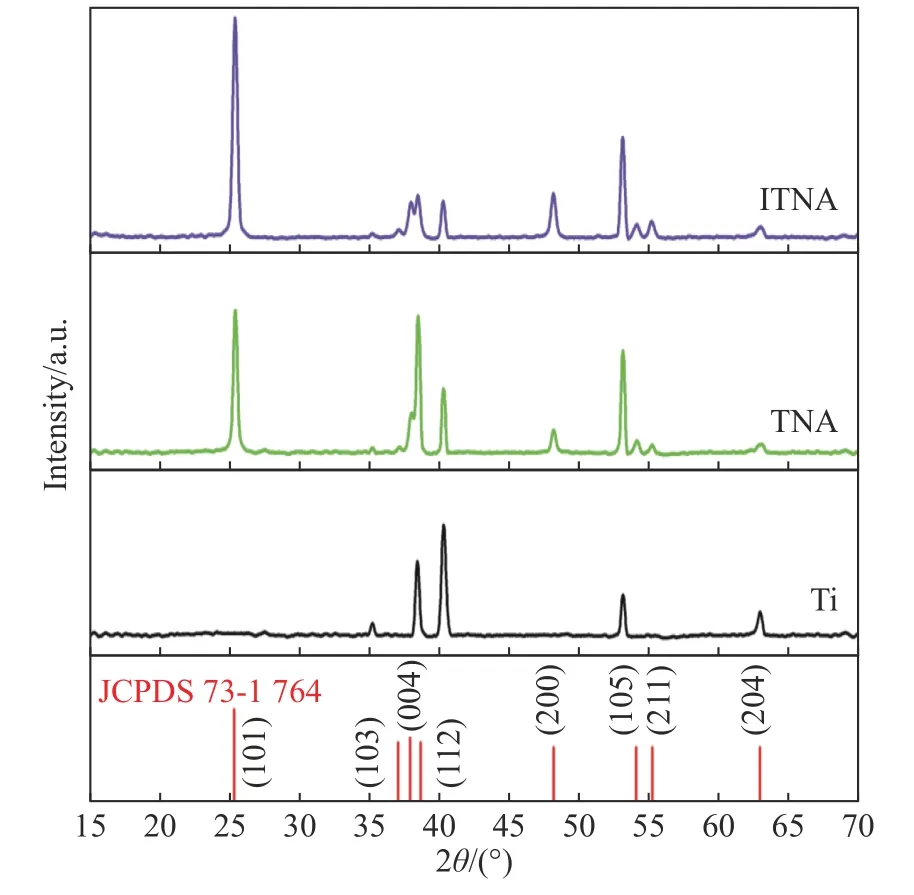
图3 Ti片、TNA 和ITNA 的XRD图谱Fig. 3 XRD patterns of Ti, TNA, and ITNA
2.2 光催化性能
2.2.1 电池结构对MB降解的影响
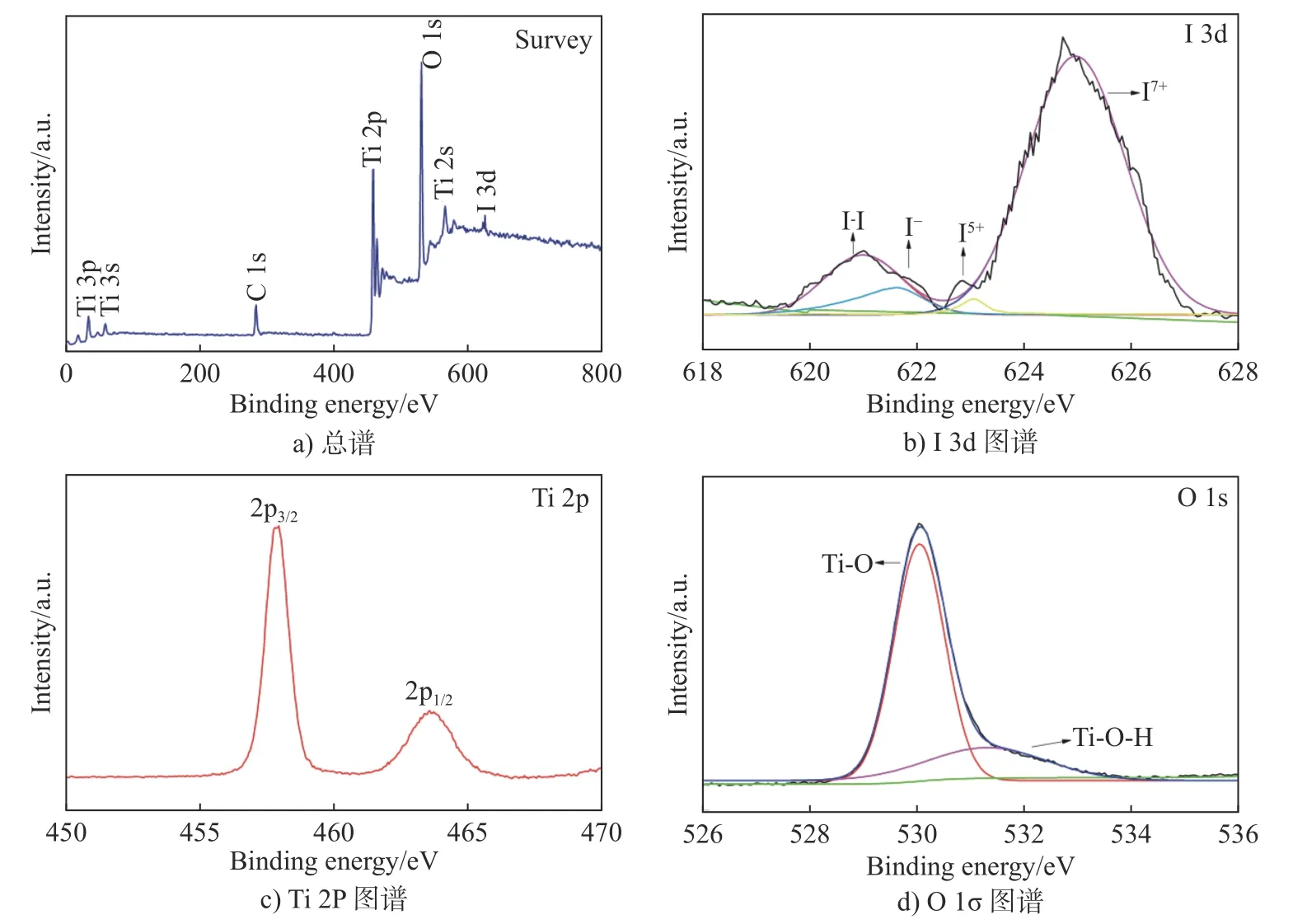
图4 ITNA的XPS图谱Fig. 4 XPS spectra of ITNA
本文使用了不同电池结构对MB的脱色情况进行研究; 其中, 阳极为ITNA, MB的浓度为6 mg·L–1, 极板间距为1.0 cm. 光氧化降解(Photo-oxidation, PO)无电极, 光催化降解(Photocatalytic,PC)仅含有TNA与ITNA光阳极.如图5a)所示, 120 min后PO条件下的脱色率为6.6%, 主要是由光降解导致、无光生电子–空穴对产生; TNA条件下脱色率为15.0%, 降解效果并不明显; ITNA条件下的脱色率达到了44.8%, 表明I掺杂明显加速了MB的降解[29], 脱色率是TNA条件下的3倍左右; f-PFC的两种结构对MB的脱色率分别达到了71.6%和66.3%; p-PFC结构对MB的脱色率达到了86.7%, 优于其他结构.

图5 在PO、TNA、f-PFC-1、f-PFC-2和p-PFC条件下对MB进行降解Fig. 5 Degradation of MB under PO, TNA, f-PFC-1, f-PFC-2, and p-PFC

采用一级动力学模型拟合了MB的脱色过程(见公式(1)),其中,ct为某个时间点的MB浓度(mg·L–1),c0为MB的初始浓度(mg·L–1),t为降解时间(min),k为脱色速率常数(min–1). 通过计算, 得到MB在PO、PC、p-PFC、f-PFC-1和f-PFC-2条件下的脱色速率常数分别为0.6 × 10–3、1.4 × 10–3、5 × 10–3、10.5 × 10–3、9.1 × 10–3和16.5 × 10–3min–1, 如图5b)所示. 在f-PFC-1中, 光照是从阴极一侧垂直射向阳极, 且阴阳极面积比为1∶3, 导致阴极遮挡了部分光照, 进而使得光子吸收减少, 活性粒子数减少; f-PFC-2中, 纳米管一侧能够降解的MB有限, 阴极与阳极中间的MB不能很快扩散到纳米管一侧, 阳极产生的活性粒子不能快速地扩散到阴极与阳极中间, 因此传质速率限制了脱色的快慢.
2.2.2 p-PFC极板间距的影响
在MB的降解过程中, p-PFC的光阳极发生下列反应:

光阳极产生的e–会通过外电路流向阴极, 产生的H+会从溶液中从阳极到达阴极, 并在阴极发生下列反应:

因此, 可以得知, 在溶液中对MB的降解起作用的主要是h+、·OH、其他氧化粒子(other oxidation species, OOS)以及MB自身的光敏化降解(self sensitization oxidation, SSO). 反应式(b)中产生的H+转移到阴极, 会促进反应式(d)和反应式(e)的正向反应进行, 进而产生更多的·OH. 由于产生的·OH寿命极短, 在水中为纳秒级[30], 由此可以推断,在p-PFC中, 阴阳极极板之间的间距会影响溶液中的传质过程, 从而影响MB的降解速率.
图6所示是p-PFC中阴阳极极板在不同间距下MB的脱色情况, MB溶液浓度为6 mg·L–1. 从图中可知, 当极板间距为1.0 cm时脱色率达到最大, 为91.7%, 并且脱色速率常数随着极板间距的增大呈现先增大后减小的趋势, 这主要与光催化反应过程的先吸附后降解的性质相关(见2.2.3节): 当间距较小的时候, 由于阳极表面MB的反应产物堆积, 使得H+的扩散受阻, 活性粒子的产生速率变慢,从而影响MB的降解, 而当距离较大的时候, H+的扩散路径变长, 产生较大的传质阻力, 导致降解速率变小[31-32]; 在极板间距为1 cm的时候, 活性粒子的产生速率和MB的反应产物扩散速率可达到一个最佳的比例, 具有最佳的脱色速率. 在本实验中的p-PFC系统中, 较为合适的阴阳极间距为1.0 cm.
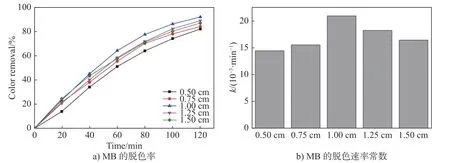
图6 p-PFC中不同极板间距对MB脱色率的影响Fig. 6 Degradation of MB with different plate spacing in p-PFC
2.2.3 MB的p-PFC降解机理
在PC过程中有机物的降解主要发生在催化剂的表面[33], 因此, 推断在p-PFC中MB的降解同样发生在ITNA的表面. MB浓度对其降解具有一定的影响, 图5所示为p-PFC对不同浓度的MB的降解情况, 极板间距为1.0 cm, 光阳极为ITNA. 图7所示为脱色率的变化, 内嵌图为不同MB浓度的脱色速率常数, 根据图示内容可以得到, MB浓度为6 mg·L–1时, 脱色率和脱色速率常数高于其他浓度, 120 min时达到了93.1%, 并且MB脱色率随浓度的增加先增大后减小.
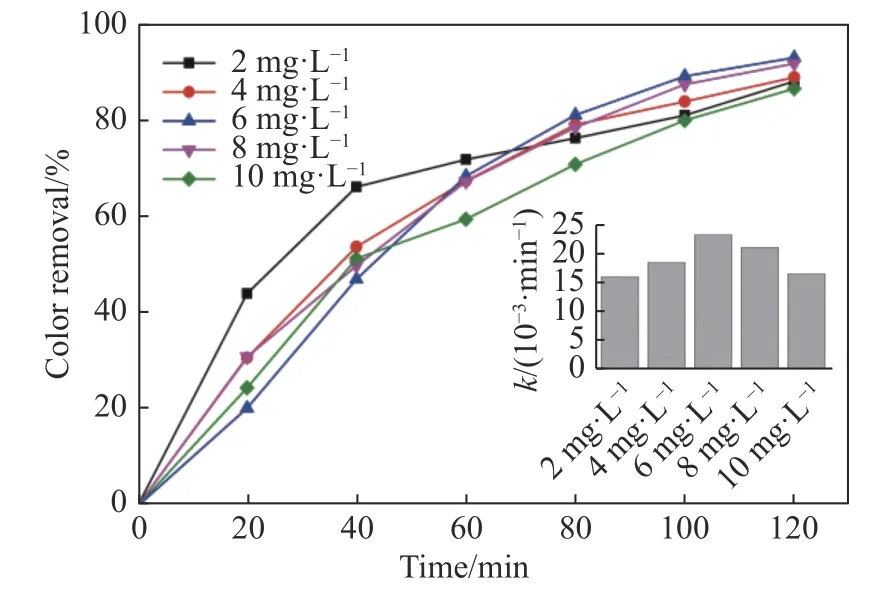
图7 p-PFC不同MB浓度对脱色率的影响Fig. 7 Percentage of color removal with different MB concentration in p-PFC
使用Langmuir-Hinshelwood (L-H) 动力学模型(见公式(2))对界面催化反应过程进行分析:
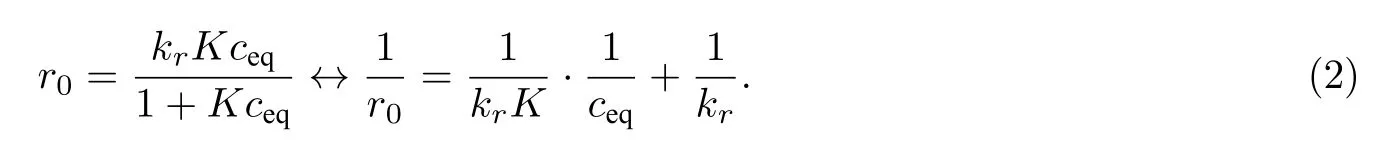
公式(2)中,r0为初始反应速率(mg·min–1·L–1),ceq为吸附平衡浓度(mg·L–1),kr为催化剂的本征降解速率常数(mg·L–1·min–1),K为催化剂的吸附常数(L·mg–1). 从图8可以看出,ceq与r0满足L-H动力学模型, 计算得到的K和kr分别为0.009 78 L·mg–1和0.813 mg·L–1·min, 证明在ITNA表面对MB的吸附是限速步骤[33]. 较好的线性关系也表明MB的降解过程主要发生在电极的表面, 是界面催化降解反应. 当浓度较低时, 表面活性位点较多, 但是分子碰撞减少, MB扩散变慢, 反应速率降低; 当浓度较高时, 光催化剂的活性反应位点有限, 吸附速率达到最大, 反而使其脱色率下降.
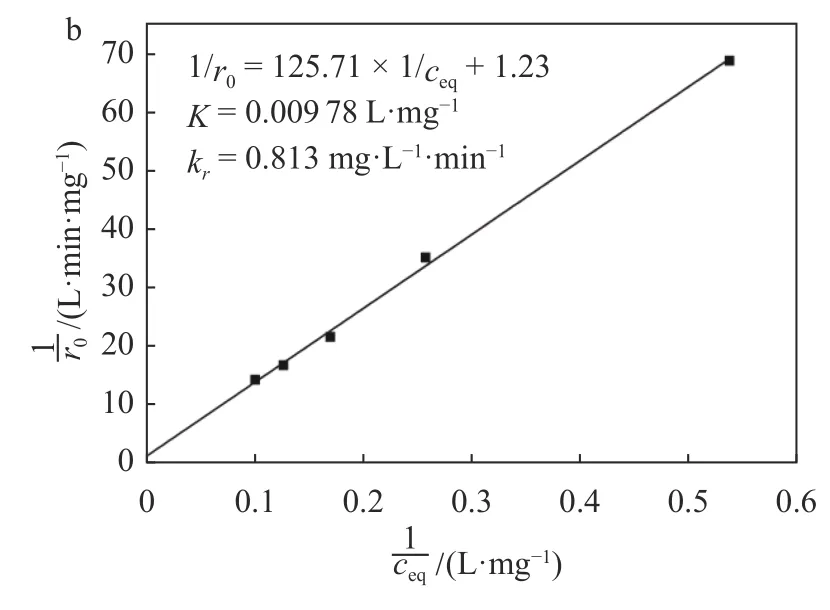
图8 MB的脱色速率常数Fig. 8 Decolorization rate constants for MB
在PFC中产生的活性粒子对MB的降解起到了重要的作用, 由反应式(a)—(e)可知, MB可以被h+、·OH、SSO、OOS降解, 因此选取草胺酸(ammonium oxalate, AO)和叔丁醇(tert-butanol, TB)分别作为h+和·OH的捕获剂, 来分析对比不同活性粒子在p-PFC与两种f-PFC结构中对MB的降解所占百分比. 如图9所示, 3种结构中, 同时添加AO和TB时对MB的降解抑制作用最为明显, 表明在MB的降解过程中h+、·OH起到了主导作用. ·OH的产生主要是由h+反应生成(见反应式(b))和H2O2自由基(HR)转化生成(见反应式(e)), 分别记为·OH(h+)和·OH(HR). 根据脱色速率常数的对比,计算出了h+、·OH(h+)、·OH(HR)、SSO、OOS占MB脱色的百分比, 如表1所示. 从表中可以看出, 在p-PFC中, 24.2%的MB被h+直接氧化, 还有20.4 %的MB被h+产生的·OH氧化, 而h+产生的H+传质到阴极也促进了HR反应产生·OH, 这说明在p-PFC中h+是引起MB脱色的最主要因素, 并且传质过程迅速, 阻碍较少. 在f-PFC-1结构中, 由h+产生的·OH明显下降, 而OOS的占比明显上升, 可能是由于光阳极部分被遮挡, 产生的h+较少. 在f-PFC-2中, 38.5% MB被h+直接降解, 光阳极只与极少部分液体接触, H+需要绕过光阳极扩散到阴极反应, 传质受阻, 由HR产生的·OH减少, 并且由于·OH寿命极短[30], 还未及时地扩散到阳极与阴极之间的区域参与MB的降解便自我消除了, 因此传质过程极大限制了MB的降解.

图9 添加h+、·OH捕获剂条件下MB的降解Fig. 9 The degradation of MB with the addition of h+ and ·OH scavengers

表1 不同PFC结构中h+、·OH(h+)、·OH(HR)、SSO和OOS占MB脱色的百分比Tab. 1 The percentages of h+, ·OH(h+), ·OH(HR), SSO and OOS in MB decolorization with different PFC structures%
2.2.4 对不同有机物的降解
对f-PFC-1、f-PFC-2、p-PFC 3种结构进一步进行了对比, 使用罗丹明B(Rhodamine B, RhB)、苯酚、甲基橙3种有机物分别进行了降解, 如图10所示. 有机物浓度均为10 mg·L–1, 极板间距为1.0 cm.从图中可得到120 min后f-PFC-1、f-PFC-2、p-PFC对RhB的降解分别达到了61.4%、59.0%、72.1%,对苯酚的降解达到了48.8%、45.7%、60.0%, 对甲基橙的降解达到了42.2%、51.0%、60.5%, p-PFC的降解性能都优于其余两种结构. 从图10d)也可看出, 降解不同有机物时, p-PFC的降解速率常数都高于f-PFC-1和f-PFC-2.
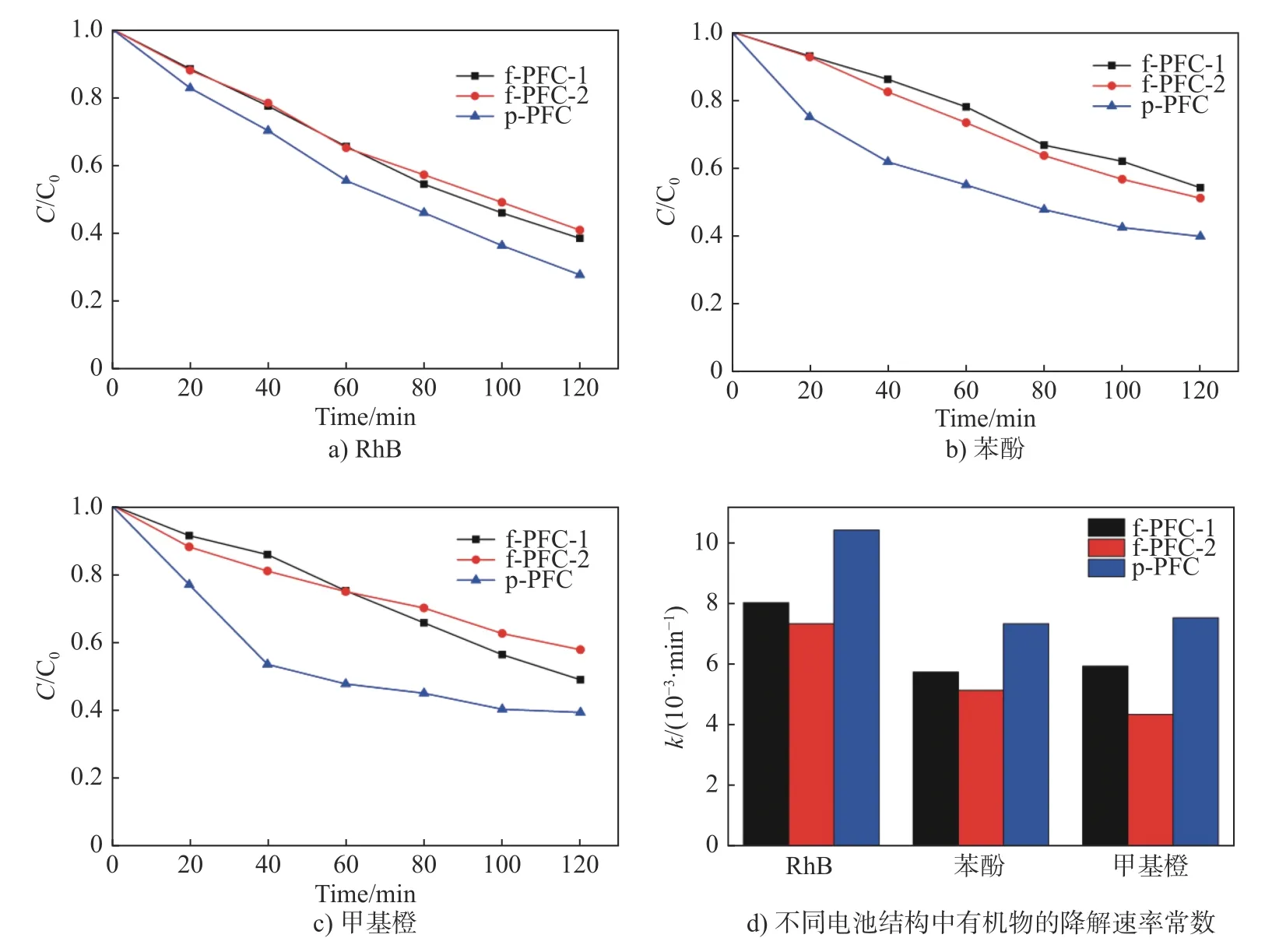
图10 不同电池结构中3种有机物的降解Fig. 10 Degradation and the decolorization rate constants of three organics in different cell structures
3 结 论
本文采用阳极氧化法制备了ITNA光阳极, 通过FESEM证实了I掺杂对TNA的形貌无明显影响, 为规则的纳米管阵列, 通过XRD、XPS证实I成功掺杂进了TNA中, 该电极表现出比TNA更加优异的降解性能. 将ITNA与Pt组合成p-PFC, 在阴阳极间距为1.0 cm、MB浓度为6 mg·L–1时降解率达到了最大, 为93.1%. 在p-PFC中, MB的降解主要发生在ITNA电极表面, 为限速步骤, 并且h+和·OH在MB的降解中发挥着主导作用. 对f-PFC-1、f-PFC-2、p-PFC 3种结构进行对比, 在降解MB时, p-PFC中h+和·OH的产生和传质效率都优于f-PFC; 降解RhB、苯酚、甲基橙时, p-PFC的降解效率也都高于f-PFC结构.
猜你喜欢
Advances in Atmospheric Sciences(2022年6期)2022-04-02 05:29:02
石油管材与仪器(2020年5期)2020-11-05 02:36:06
电子制作(2018年12期)2018-08-01 00:47:46
中学生数理化·高二版(2016年10期)2016-12-24 11:26:42
湖南农业(2016年3期)2016-06-05 09:37:36
锻压装备与制造技术(2016年3期)2016-06-05 09:36:12
电源技术(2015年9期)2015-06-05 09:36:06
电源技术(2015年9期)2015-06-05 09:36:06
电源技术(2015年9期)2015-06-05 09:36:04
特产研究(2014年4期)2014-04-10 12:54:18
