基于流式细胞术和基因组survey分析的地黄基因组研究
2021-02-03 07:45朱畇昊韩永光马利刚冯卫生郑晓珂
中草药 2021年3期
赵 乐,朱畇昊,王 敏,韩永光,马利刚,冯卫生,郑晓珂*
1.河南中医药大学药学院,河南 郑州 450046
2.呼吸疾病诊疗与新药研发河南省协同创新中心,河南 郑州 450046
3.北京工商大学 化学与材料工程学院,北京 100048
地黄Rehmannia g lutinosaLibosch 是玄参科地黄属多年生草本植物,为《中国药典》2015年版药材地黄的唯一来源植物[1],是我国传统的大宗中药材,具有很高的药用价值和经济价值。地黄以其块根入药,块根中含有环烯醚萜、黄酮、木脂素及多糖等多种化合物[2]。地黄药理活性显著,对心脑血管系统、神经系统、免疫系统均有一定保护作用[3]。梓醇是地黄的主要活性成分,从化学结构分析,梓醇属于环烯醚萜苷类化合物,具有降血糖、神经保护、抗炎、抗氧化等多种生物学活性[4]。Yan 等[5]发现梓醇能够缓解2 型糖尿病中的胰岛素抵抗,起到降血糖的作用,还能减缓糖尿病晚期的并发症。此外,梓醇还可以改善小鼠阿尔茨海默症的病理特征,同时改善相关的行为障碍[6-7]。
目前关于地黄的研究,主要集中于化学成分的分离、梓醇和地黄其他活性成分的药理作用、以及地黄连作障碍等方面,关于地黄基因组方面的研究报道[8]较少。Zeng等[9]对地黄属6种地黄植物的叶绿体基因组进行了测序,同时进行了系统发育关系分析,发现地黄叶绿体基因组大小为153 622 bp,而且地黄属6 种地黄的叶绿体基因组在基因结构方面高度一致。闫坤等[10]通过观察地黄属6 种地黄的小孢子母细胞减数分裂,发现地黄和茄叶地黄属于四倍体植物(2n=56),天目地黄、高地黄、裂叶地黄和湖北地黄属于二倍体植物(2n=28),说明地黄属植物存在多倍体现象。这种多倍体现象可能是由于全基因组复制形成的,王姣等[11]利用系统发育生物学方法,通过分析地黄属5 种地黄的转录组数据,发现地黄属植物经历了2 次全基因组复制,加速了地黄属植物的物种分化,通过二倍体祖先的基因组加倍形成了四倍体植物。闫婧[12]用二代高通量测序对地黄属的天目地黄基因组进行了survey 分析,发现天目地黄基因组大小估算为1.14 Gb,GC 含量为40.64%,重复序列比例为 76.94%,杂合度为0.34%,从基因组基本结构特征分析,天目地黄基因组属于高重复、低杂合的大型基因组。
地黄的许多栽培种在长期的栽培过程中受到了选择,这些栽培品种之间的遗传关系不清楚,而且这些栽培品种在叶和花的颜色和形态、以及根的形状方面变异较大[13]。由于地黄栽培品种之间的遗传关系不清楚,因此获得地黄的全基因组精细图谱对于研究其主要药用成分—梓醇的生物合成及代谢调控途径具有重要的意义,同时也为研究地黄株高、叶形、块根膨大等性状变异奠定了基础。根据报道地黄为四倍体(2n=56)[10,13],本研究以地黄栽培品种“沁怀1 号”为材料,通过二代高通量测序技术对地黄基因组进行survey 分析,同时结合流式细胞技术来评估地黄基因组大小,最终获得地黄全基因组重复序列比例、基因组杂合度以及GC 含量等信息。这些信息将为今后绘制地黄全基因组的精细图谱提供依据,也为阐明梓醇的生物合成及代谢调控途径奠定基础。
1 材料
地黄所用材料为地黄栽培品种“沁怀1 号”组培苗,经河南中医药大学董诚明教授鉴定为地黄Rehmannia glutinosaLibosch,以已知基因组大小的大豆组培苗(中国农业科学院)和辣椒组培苗(中国农业科学院)作为流式细胞技术测定的对照样品,采集这3 种植物组培苗新鲜的叶片,经液氮速冻后,放置于-80 ℃超低温冰箱保存。
BD FACSCalibur 流式细胞仪(美国BD 公司),离心机5810R(德国Eppendorf 公司);碘化丙啶(propidium iodide,PI)批号340242,购自美国BD公司;吐温20(批号P2287)购自美国Sigma-Aldrich公司。Otto I 缓冲液:0.1 mol/L 柠檬酸、0.5%聚山梨酯20,用0.22 μm 滤器滤过后4 ℃保存。Otto II缓冲液:0.4 mol/L Na2HPO4·12H2O,用0.22 μm 滤器滤过后室温(18~25 ℃)保存。
2 方法
2.1 细胞核悬液制备及基因组大小测定
根据Doležel 等[14]的方法,各取地黄、大豆和辣椒组培苗新鲜叶片100 mg,放入培养皿中,加入1 mL 预冷的Otto I 缓冲液,用锋利的刀片将组织切碎,用移液器上下吹打几次混合均匀(注意避免气泡),然后将匀浆液通过42 μm 尼龙筛网滤过到离心管中,4 ℃、1000 r/min离心5 min,小心的弃去上清,收集细胞核沉淀。再用1 mL Otto II 缓冲液重悬细胞,然后加入DNA 荧光染料碘化丙啶(PI)和 RNA 酶,使二者的终浓度均在 50 μmol/mL,置于冰上,避光染色20 min,再将地黄、大豆和辣椒的染液混合形成混合样品,然后上机测定这3 种植物的单独样品和混合样品。
将已知基因组大小的大豆和辣椒作为对照样品,首先用流式细胞仪单独测定地黄、大豆和辣椒基因组DNA 的相对荧光强度,然后检测混合样品的相对荧光强度,并根据不同样品相对荧光强度峰值的大小,同时参考对照样品的基因组,计算地黄的基因组大小。用流式细胞仪测定时,每个样品收集10 000 个颗粒,检测PI 的荧光强度,收集FL2 通道的荧光,使用FACSComp 软件对数据进行分析和处理,并通过FL2-A通道计算得到每个样品G1 期的荧光强度[15]。
2.2 地黄基因组survey 分析
采用改良CTAB 法提取地黄叶片的基因组DNA,用紫外分光光度计和琼脂糖凝胶电泳分别检测地黄基因组DNA 的浓度、纯度和完整性。
检测合格后,用超声波震荡方法将地黄基因组DNA 破碎至目的片段(350 bp),然后经过末端修复、加A、加接头、目标片段选择和PCR等步骤构建小片段测序文库;用安捷伦2100 和定量PCR检测文库片段大小和文库定量,确定文库是否符合测序标准;通过桥式PCR 将文库固定到测序芯片上;利用北京百迈客Illumina Hiseq 4000 测序仪对构建好的文库进行双端150 bp(PE 150)测序,测序所产生的数据经过质控后用于下一步信息分析。双端测序数据经过评估(GC 分布统计、质量值Q20、Q30评估)、过滤后得到高质量的数据,用于地黄基因组大小的评估、基因组的组装、GC 含量的统计、杂合度的统计(以及组装后的评估)。根据地黄基因组大小,结合流式细胞技术测定结果来估算测序深度。
Kmer 是从测序数据中提取出的长度为K的寡聚核苷酸序列,本研究中取K=19 进行分析,在测序reads 均匀分布的前提下,根据公式,计算基因组大小(基因组大小=总碱基数/平均测序深度=总Kmer 数/平均Kmer 深度)。由于测序片段是随机打断的,标准的Kmer 深度分布曲线呈正态分布,根据实际曲线偏离正态分布的程度,通过Jellyfish和GenomeScope 软件[16]对测序数据进行分析,统计产生Kmer 频数分布数据,再使用该数据进行拟合作图,得到Kmer 分布图,进一步评估地黄基因组杂合度和重复序列比例。
3 结果与分析
3.1 流式细胞技术测定地黄基因组大小
以已知基因组大小的物种为对照品:大豆(基因组大小为1.1 Gb)[17-18]和辣椒(基因组大小为3.2 Gb)[19],进行细胞流式实验,来估算地黄基因组的大小。首先用流式细胞技术将大豆、地黄和辣椒单独测定,检测每个样品的相对荧光强度(图1-A~C),然后采用内标法,以大豆和辣椒为对照品,用流式细胞技术对混合样品进行相对荧光强度的检测,大豆、地黄和辣椒混合样品的基因组DNA 相对荧光强度峰值分别为41.99、73.25、110.61(图1-D)。根据流式细胞实验结果,地黄基因组大小介于大豆和辣椒之间,估算地黄基因组大小应在2.00~2.12 Gb。
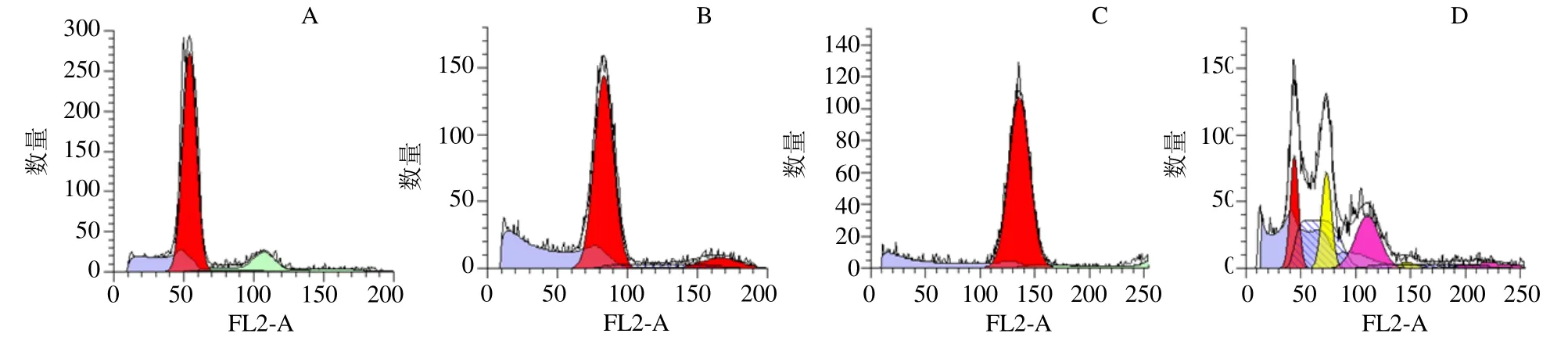
图1 不同样品流式细胞术检测结果Fig.1 Peak value image of different species
3.2 测序结果统计
使用地黄“沁怀1 号”样品的基因组DNA 构建350 bp 文库,在Illumina Hiseq 4000 测序平台测序并过滤得到132.79 Gb 高质量的数据,总测序深度约为66.40×,测序数据Q20比例均在97.15%以上,Q30比例均在92.86%以上,满足60×以上的测序数据量。
3.3 样品污染评估
如果地黄基因组DNA 样品存在污染不仅会降低有效数据量,同时还会影响基因组survey分析结果的准确性,导致基因组大小、重复序列比例、杂合度和GC 含量等基因组特征评估结果出现较大偏差,使得基因组组装建库策略出现偏差,最终影响后续的基因组组装效果。为了判断提取的地黄基因组DNA 是否受到污染,从测序得到的350 bp 文库中,随机取10 000 条单端reads,与NT 库(Nucleotide Sequence Database)进行BLAST 比对。BLAST 使用ncbi-blast+2.2.29 版本,参数设置为“-num_descriptions 100-num_alignments 100-evalue 1×10-5”。能够比对上NT 库的reads 占提取reads数的8.74%,其中比对到芝麻Sesamum i ndicumL.、宽叶沟酸浆Erythranthe guttatusDC.、可可Theobroma cacaoL.、番茄Solanum lycopersicumL.和地黄的reads 数分别占比对上NT 库reads 数的12.47%、4.57%、4.11%、3.08%和2.17%。芝麻是地黄的近缘物种,于2014年完成芝麻基因组测序[20],在NT 库中基因注释信息丰富,所以在比对物种所占比例中最高为12.47%,在前期地黄基因的进化分析中也发现地黄和芝麻的亲缘关系较近[21-24],而地黄基因组信息未知,在NT 库中基因注释极少,所以在比对物种所占比例中仅为2.17%。比对结果中未发现动物、微生物等异常比对,因此该地黄基因组DNA 样品测序数据不存在污染,可用于基因组survey分析。如果有一定比例的reads 比对到进化距离较远的物种如动物、微生物等,则判断样品可能存在污染,需要进一步检查原因,不能用于基因组survey 分析。
3.4 核外DNA 含量评估
由于细胞核外也存在核酸序列,如果测序文库中核外DNA 含量过高,会加大后期基因组组装的难度,甚至产生错误。因此评估文库中核外DNA 含量对判断数据能否用于后续基因组组装非常必要。为了评估测序数据中核外DNA 的含量,利用Illumina Hiseq 测序得到的350 bp 文库与地黄的叶绿体序列(NC_034308.1,153 622 bp)进行SOAP 比对[25]。SOAP 使用2.21 版本,参数设置为“-m 260-x 440”。双端比上的reads 数为17 415 302,占总reads 的1.96%,单端比上的reads 数为11 313 201,占总reads 的1.28%,这2个的比例都低于经验值5%。由此判断测序数据的核外DNA 含量很低,不影响基因组survey 分析的准确性和后期基因组的组装。
3.5 地黄基因组大小、重复序列比例和杂合度评估
使用350 bp 文库数据构建K=19 的Kmer 分布图(图2),进行地黄基因组大小、重复序列比率和杂合度的评估。由图2 可观察到明显的3 个峰,第1个峰的Kmer 深度分布为27,第2 个峰的Kmer 深度分布为56,第3 个峰的Kmer 深度分布为102,这3个峰的Kmer深度分布符合1∶2∶4的趋势,推测地黄为四倍体。假设地黄基因组为“AABB”,则代表“AA”一致,“BB”一致,但“AA” “BB”不同,根据地黄基因组呈现的Kmer 分布,从测序数据中得到的总Kmer 数为116 809 179 227 个,去除深度异常的Kmer 后,共113 026 782 538 个Kmer用于基因组大小估计,用Jellyfish 和GenomeScope软件计算得到的基因组大小约2.03 Gb,与流式细胞技术测定结果一致。根据Kmer 分布情况,估计地黄基因组重复序列比例约 78.48%,杂合度约1.93%。
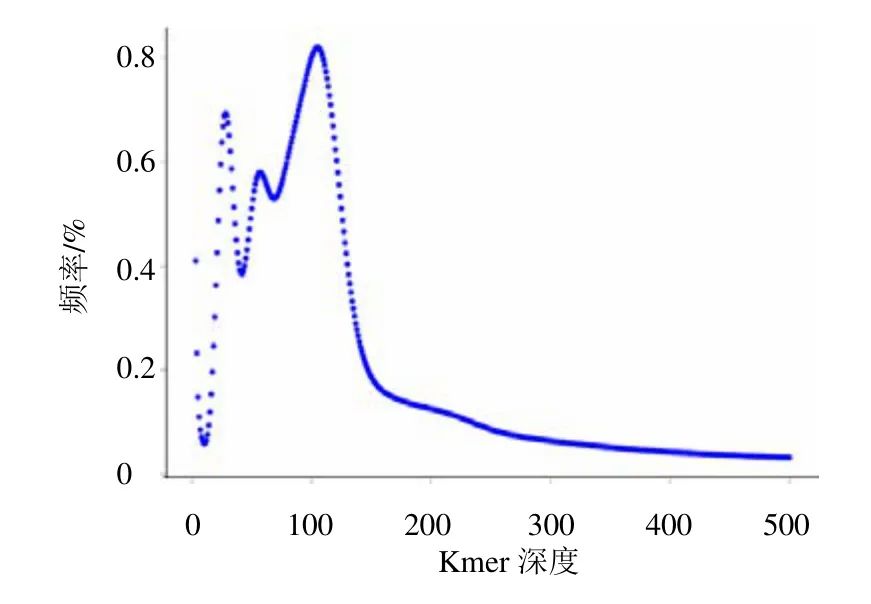
图2 Kmer 分布图预估地黄基因组大小Fig.2 Kmer analysis for estimating genome size of R.glutinosa
基因组GC 含量对二代基因组测序的随机性有较大影响,过高(>65%)或过低(<25%)的GC含量会导致测序偏向性,严重影响基因组分析结果。物种GC 含量是评估基因组survey 分析准确性和后续基因组组装难度的重要指标之一,通过对350 bp 文库测序数据分析,地黄基因组的GC 含量约37.27%,较为适中,不会影响分析的准确性。
综上所述,地黄基因组大小约2.03 Gb,重复序列比例约78.48%,杂合度约1.93%,基因组的GC 含量约37.27%,从基因组基本结构特征分析,地黄基因组属于高重复、高杂合、大基因组的复杂基因组。
4 讨论
基因组是一个生物体中全部的DNA 信息,包括其所有基因,通过对药用植物的全基因组进行测序,获得药用植物基因组的DNA 序列信息,就能够从分子水平研究活性成分的生物合成和代谢调控途径,以及药用植物之间的进化关系。随着高通量测序技术的迅速发展,目前已有几十种药用植物的全基因组被解析,如大麻、灵芝、铁皮石斛、丹参、三七、罂粟、黄花蒿等重要的药用植物[26]。这些药用植物全基因组序列信息的发布,为药用植物活性成分生物合成和代谢调控途径的研究奠定了坚实的基础,极大的推动了药用植物分子生物学的研究水平,也为药用植物的遗传育种提供了重要依据[27]。
由于流式细胞技术方便、快捷而且结果可靠,是测定植物基因大小的首选方法[14],目前应用流式细胞技术,测定了黄芪[28]、柴胡[29]和人参[30]等药用植物基因组的大小。除了流式细胞技术以外,利用高通量测序技术对植物基因组进行survey 分析,也是检测植物基因组大小和特征的有效方法。通过对基因组进行survey分析,提前获得基因组的大小、重复序列、GC 含量等信息,为后续进行全基因组深度测序时选择合适的测序策略提供依据。目前利用高通量测序方法,已对罗汉果[31]、马鞍藤[32]、三岛柴胡[33]等药用植物的基因组进行survey分析,获得了这些植物基因组大小、复杂程度以及基因组的其他特征等信息。
玄参科有4500 种植物,包括多种药用植物,如地黄、玄参、洋地黄、胡黄连、阴行草等,具有重要的药用价值和经济价值[34],但是对于这些药用植物进行遗传育种和基础研究的主要障碍在于缺乏参考基因组序列,截至目前还没有玄参科植物的参考基因组发布。准确测定地黄基因组大小是后续进行地黄全基因组深度测序和遗传分析的基础,目前在许多基因组测序项目中都使用高通量测序的Kmer 分析估算基因组大小[35]。本研究应用流式细胞技术估算地黄基因组大小应在2.00~2.12 Gb,同时结合高通量测序的Kmer 分析,地黄基因组大小修正为2.03 Gb,而且通过Kmer分析还能获得地黄基因组的重复序列78.48%、杂合度1.93%、GC 含量37.27%等基因组特征信息。通过将流式细胞技术和基因组survey分析2种方法结合在一起,对地黄基因组大小和特征进行分析,提高了实验结果的可靠性。从基因组基本结构特征上看,地黄基因组属于高重复、高杂合、大基因组的复杂基因组。由于二代高通量测序技术限制,测序读长太短,仅有200~500 bp,对于地黄这种大型复杂基因组的组装难度大、花费多,而从头组装de novo组装对算法的要求更高,所以针对地黄基因组杂合度高、重复序列比例高、基因组大的特点,可采用3 代高通量测序技术(PacBio Sequel)同时结合染色质区域捕获(chromosome conformation capture,Hi-C)技术来完善三代高通量测序基因组组装结果,将其组装到染色体水平[36]。PacBio Sequel 测序平均长度达12~15 kb,能够跨越基因组高重复区域和高复杂区域,减少拼接成本,同时结合Hi-C 技术,能够获得高质量染色体水平的基因组序列,可以阐明物种间基因组的演化历程,揭示物种间的进化关系。本研究通过流式细胞技术和基因组survey 分析,获得的地黄基因组大小和特征等信息,为今后绘制地黄全基因组的精细图谱奠定了基础,也为研究地黄属植物的多倍体现象以及遗传进化关系提供依据。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
中国现代中药(2022年3期)2022-04-11
中国现代中药(2021年9期)2021-11-16
猪业科学(2021年3期)2021-05-21
种子(2021年3期)2021-04-12
幽默大师(2020年10期)2020-11-10
中华诗词(2019年1期)2019-11-14
特产研究(2019年3期)2019-09-21
猪业科学(2018年4期)2018-05-19
少儿科学周刊·儿童版(2017年5期)2017-06-29
外语教学理论与实践(2016年1期)2016-06-11