吉西他滨单磷酸盐/紫杉醇联用靶向纳米粒的制备及其大鼠体内药动学研究
2021-02-03 07:44赖梦琴
中草药 2021年3期
赖梦琴,张 鹏,,杨 明,,李 翔,*,张 婧,*
1.江西中医药大学 现代中药制剂教育部重点实验室,江西 南昌 330004
2.江西中医药大学 创新药物与高效节能降耗制药设备国家重点实验室,江西 南昌 330006
3.九江学院 临床医学院/附属医院,江西 九江 332000
紫杉醇(paclitaxel,PTX)通过促进微管聚合,抑制微管蛋白解聚,诱导细胞周期阻滞在G2与M期并促进凋亡,具有独特的抗肿瘤机制[1-2],被广泛应用于乳腺癌等各种肿瘤的治疗,然而紫杉醇几乎不溶于水、且无选择性全身分布是限制其临床应用的主要原因[3-4]。为此,制剂工作者尝试采用多种制剂技术方法改善其水溶性及靶向性[5-6]。吉西他滨(gemcitabine,GEM)作为水溶性胞嘧啶核苷衍生物,是细胞周期特异性抗代谢药物,通过核苷激酶磷酸化,转化成具有活性的二磷酸及三磷酸核苷发挥抗肿瘤作用[7]。紫杉醇/吉西他滨作用机制相异、无交叉耐药性[8],联合用药后毒性显著下降[9-10],然而由于紫杉醇与吉西他滨溶解性完全相反,且吉西他滨的体内半衰期比较短,影响了药物联用的临床效果。
由于第1 步的单磷酸化反应为吉西他滨能够产生活性的重要限速步骤[11-12],因此本课题使用吉西他滨单磷酸盐(gemcitabine monophosphate,GMP)作为吉西他滨替代药物。前期研究结果表明GMP具有和吉西他滨相同的作用机制,且无明显骨髓抑制反应[13],本课题组前期已成功采用反相微乳液法在反相微乳体系中制备了尺寸均一、分散性良好且负载GMP 药物的单层脂质包裹的磷酸钙纳米核心(GMP-Cores)[14]。
为了验证并实现GMP 和紫杉醇的协同作用,本实验进一步将GMP-Cores、紫杉醇、胆固醇、长循环靶向磷脂分散于有机溶剂中,采用薄膜分散法对GMP-cores 进行二次包裹,得到非对称脂质双层修饰的紫杉醇/GMP 的纳米粒(asymmetric lipid layers coated nanoparticles co-loaded with gemcitabine monophosphate and paclitaxel,P/G-NPs,结构示意图见图1),并对所得P/G-NPs 的理化性质及体内药动学行为进行评价。
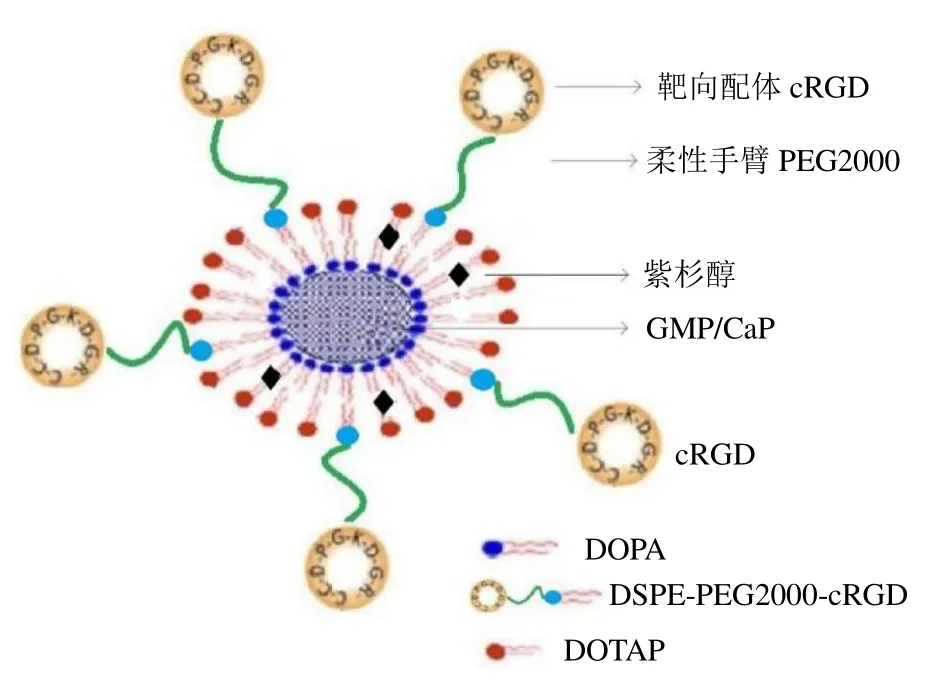
图1 P/G-NPs 结构示意图Fig.1 Schematic structure of P/G-NPs
1 仪器与材料
1.1 仪器
JEM-1200 EX 型透射电子显微镜(TEM),日本JEOL 公司;Nano ZS 型纳米粒度仪,英国Malvern公司;3-18K 型冷冻高速离心机,德国Sigma 公司;Qtrap4500 三重四级杆液质联用仪,美国AB Sciex公司;减压旋转蒸发仪,瑞士Buchi 公司;Bio-RAD 680 酶标仪,美国Bio-RAD 公司;FV1000-IX81 激光共聚焦显微镜,日本Olympus 公司;BSA124S电子分析天平,北京赛多利斯天平有限公司;KQ5200BE 型数控超声清洗器,昆山市超声仪器有限公司;JY92-ⅢN 探头超声仪,宁波新芝生物科技股份有限公司;DF-101S 磁力搅拌器,巩义市予华仪器有限公司。
1.2 材料
紫杉醇,质量分数≥97%,美国Sigma-Aldrich公司;GMP,质量分数≥97%,常州施嘉生物医药科技有限公司;紫杉醇注射剂(规格5 mL∶30 mg),批号1604201,四川三精升和制药有限公司,国药准字H20046119;多西紫杉醇(doxorubicin,DOX),质量分数≥99.0%,批号18072044,上海同田生物技术股份有限公司;头孢克洛(cefaclor,CEC),质量分数94.4%,批号130481-201205,中国食品药品检定研究院;1,2-二油酰基-3-三甲基铵-丙烷(甲基硫酸盐)( 1,2-dioleoyl-3-trimethylammoniumpropane chloride salt,DOTAP)和二油酰磷脂酸(dioleoylphosphatidic acid,DOPA),美国Avanti Polar Lipids 公司;1,2-二硬酯酰-sn-甘油-3-磷酰乙醇胺-聚乙二醇 2000(1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000],DSPE-PEG2000)和1,2-二硬酯酰-sn-甘油-3-磷酰乙醇胺-聚乙二醇 2000-cRGD(1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000]-cRGD,DSPEPEG2000-cRGD),西安瑞禧生物科技有限公司;表面活性剂聚氧代乙烯(5)壬基苯基醚,支化(Igepal®CO-520),美国Sigma 公司;实验用水为去离子水,甲醇、乙腈为色谱纯;其余试剂或药品均为分析纯。
人乳腺癌MCF-7 细胞,北京鼎国昌盛生物技术有限责任公司;RPMI 1640 培养基和胎牛血清,美国Gibco 公司;CCK-8 试剂盒、胆固醇,美国Sigma公司。SD 大鼠,体质量(200±20)g,雄性,湖南斯莱克景达有限公司提供,合格证号:SCXK(湘)2019-0004。所有动物实验遵循江西中医药大学有关实验动物管理和使用的规定,均符合3R 原则。
2 方法与结果
2.1 体外协同作用考察
取对数生长期的MCF-7 细胞,以细胞数5×104/mL 接种在96 孔板中,于37 ℃、5% CO2培养箱中孵育24 h,弃去上层培养液,分别加入含有不同物质的量比药物溶液的培养基(free PTX/GMP,P/G-Free)(给药组为实验组),同时设置阴性对照孔(以未给药的含细胞组为对照组)和空白孔(RPMI 1640 培养基的不含细胞孔作为空白组),每个浓度设3 复孔,24 h 后每孔加入CCK-8 液10 μL,混匀,37 ℃继续培养2 h,以酶标仪检测450 nm 处的吸光度(A)值,按照说明书计算细胞生长抑制率[生长抑制率=(A对照-A实验)/(A对照-A空白)]。P/G-Free对MCF-7 细胞的生长抑制率结果见表1。
采用金正均Q值法判断两药是否具有协同作用,计算q值[15]:q=Eab/(Ea+Eb-Ea×Eb),其中Ea、Eb为单药抑制率,Eab为两药合用抑制率。
q值计算结果见表2,其中,q>1.15 为协同作用,0.85≤q≤1.15 为相加作用,q<0.85 为拮抗作用。结果见表2,当GMP 与紫杉醇混合物质的量比为17∶1 时,q值为0.908,表现为相加作用且抑制率最高。
2.2 P/G-NPs 的制备
2.2.1 GMP-Cores 的制备 采用反向微乳法制备纳米核心GMP-Cores[14]。取一定量含有环己烷-Igepal®CO-520 溶液(71∶29)的混合物作为油相,分成2份(A 和B)。A 油相中滴加300 µL 60 mmol/L CaCl2溶液;B 油相中滴加300 μL 由75 μL 50 mmol/L Na2HPO4、90 μL 60 mmol/L GMP 组成的水溶液,搅拌均匀后加入1 mL 20 mmol/L DOPA,然后将A、B 2 相混合,继续在混合液中滴加1 mL 20 mmol/L DOPA,20 min 后待反应完成,加入等量无水乙醇破乳,4 ℃高速离心(10 000×g)20 min,弃去上清液,无水乙醇洗涤2 次后,将得到的GMP-Cores溶解在氯仿中,置于-20 ℃冰箱中备用。

表1 P/G-Free 对MCF-7 细胞的生长抑制率 (n = 3)Table 1 Inhibition rates of P/G-Free on MCF-7 cells (n = 3)
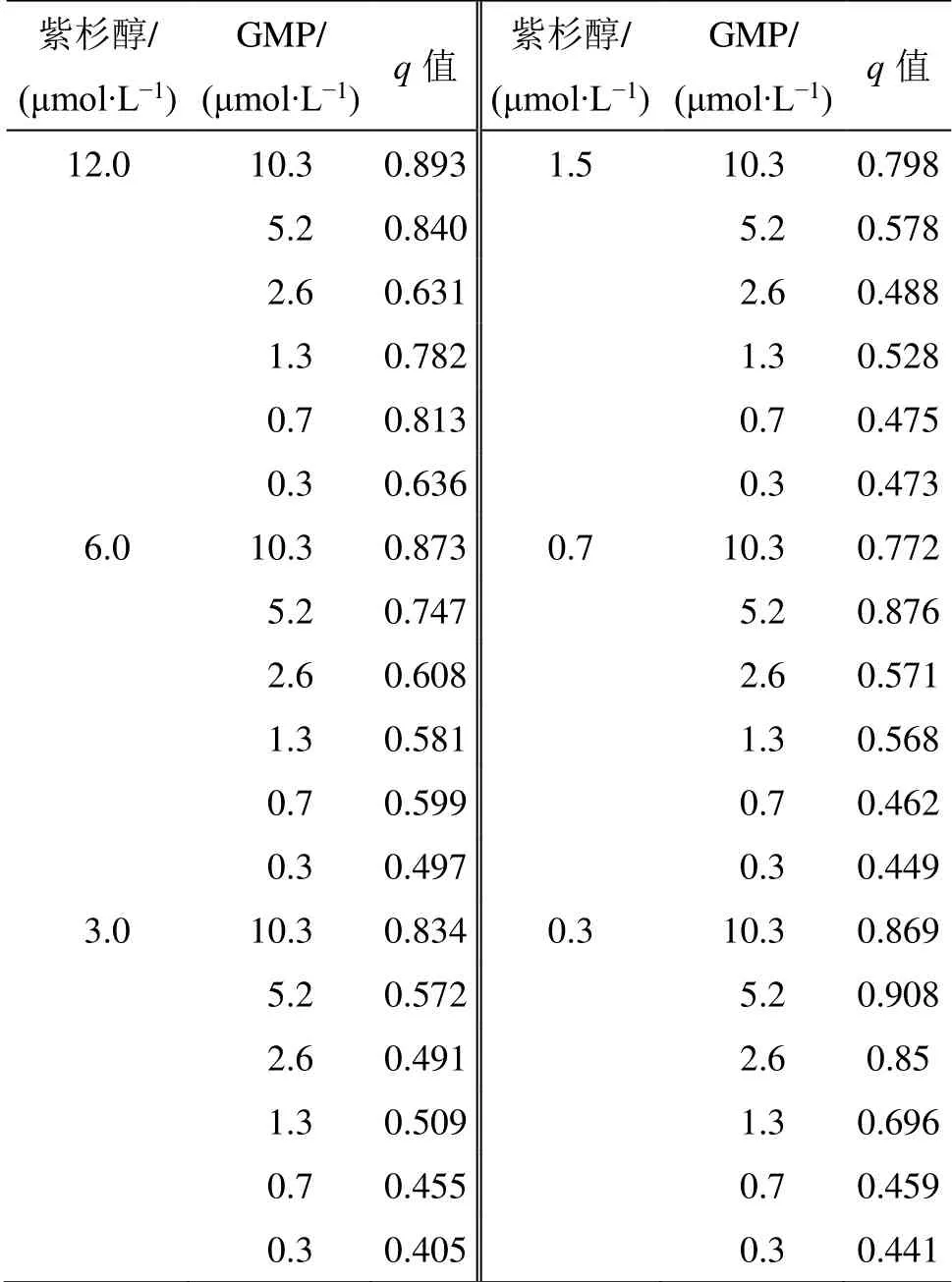
表2 P/G-Free 对MCF-7 细胞的协同抑制作用 (n = 3)Table 2 Synergistic inhibition effect of P/G-Free on MCF-7 cells (n = 3)
2.2.2 P/G-NPs 的制备 采用改进的薄膜分散法[16],制备P/G-NPs。将GMP-Cores 与紫杉醇(GMP 与紫杉醇的物质的量比为17∶1),总磷脂与胆固醇质量比为 7.1∶1,含 cRGD 多肽的磷脂混合物(DOTAP、DSPE-PEG2000、DSPE-PEG2000-cRGD质量比3.5∶11∶1)共同溶解于一定量氯仿中,旋转真空蒸发除去有机溶剂,并在恒定的氮气流下进一步干燥30 min,以在玻璃壁表面形成含药的脂质薄膜。以水为水化介质,60 ℃水浴搅拌30 min,洗脱脂质薄膜,所得混悬液于200 W 功率下探头超声2 min,得到的混悬液通过0.22 μm 滤膜滤过,并储存在4 ℃。
2.3 理化性质考察
2.3.1 GMP 和紫杉醇的测定 采用三重四级杆液质联用仪(LC-MS/MS)测定GMP 和紫杉醇的含量。
(1)色谱条件:色谱柱为Welch Materials 柱(50 mm×2.1 mm,1.8 μm);流动相为乙腈-0.1%甲酸水溶液,梯度洗脱:0~1 min,50%乙腈;1~2.5 min,55%乙腈;2.5~3 min,96%乙腈;3~6.6 min,10%乙腈;体积流量0.28 mL/min;自动进样器温度25 ℃,进样体积10 μL。
(2)质谱条件:质谱仪在多离子反应监测模式下以正离子模式操作。用于GMP 和内标CEC 的定量分析的离子反应比分别为m/z342.0→301.2和m/z368.1→174.1。GMP 与内标的碰撞能量分别为15、16 eV。紫杉醇和内标DOX 的定量分析的离子反应比分别为m/z876.3→308.1 和m/z830.5→549.3。紫杉醇和内标的碰撞能量分别为37、36 eV。电离条件包括使用电喷雾离子源,注入电压为5.5 kV,离子源温度为600 ℃,GS1 和GS2 压力为379.21 kPa(55 psi),碰撞气体压力为48.26 kPa(7 psi)。
以质量浓度为纵坐标(Y),药物峰面积与内标峰面积比值为横坐标(X)建立标准曲线,得紫杉醇、GMP 线性回归方程分别为Y=0.113 1X+0.050 3(r=0.999 0)、Y=0.019 9X-0.003 8(r=0.998 5),线性范围依次为紫杉醇 0.02~20.00 μg/mL、GMP 0.01~10.00 μg/mL。
2.3.2 包封率与载药量的测定 采用超滤离心分离游离药物[17]。取P/G-NPs 适量置于超滤离心管中(截留相对分子质量10 000),4 ℃离心(4000×g)15 min,收集滤液,采用“2.2”项下方法测定滤液中GMP、紫杉醇含量(Wfree);另取等体积P/G-NPs,按照1∶4 的体积比加入有机溶剂[四氢呋喃-1 mol/L 盐酸(7∶3)],涡旋混匀,测定混合液中GMP、紫杉醇含量(Wtotal),计算P/G-NPs 的包封率[包封率=(Wtotal-Wfree)/Wtotal]与载药量[载药量=(Wtotal-Wfree)/W纳米粒],其中纳米粒总质量(W纳米粒)由冷冻干燥后称定计算得到。结果紫杉醇、GMP的包封率分别为(98.70±0.50)%、(93.60±1.20)%(n=3),载药量分别为(0.800±0.004)%、(6.300±0.100)%(n=3)。
2.3.3 形态及粒径、Zeta 电位测定 分别取GMPCores 和P/G-NPs 5 μL 滴加于铜网上,2 min 后吸去多余液体,自然晾干,TEM 下观察,GMP-Cores呈球形,分散性好,透射电镜测得的粒径约20 nm(图2-A)。表面修饰非对称的磷脂双分子层,并将脂溶性的紫杉醇嵌入于磷脂双分子层后,观察到球形或椭球形P/G-NPs(图2-B)。

图2 GMP-Cores (A) 和P/G-NPs (B) 的TEM 图Fig.2 TEM of GMP-Cores (A) and P/G-NPs (B)
采用马尔文激光粒度测定仪测定其粒径及Zeta电位。结果P/G-NPs 的平均粒径为(85.7±10.5)nm(n=3),多分散指数为0.14±0.06(n=3),Zeta电位为(18.3±0.63)mV(n=3)。
2.4 体外释放度考察[18]
采用流通池法考察P/G-NPs 的体外释放(CE7 Smart,美国Sotax 公司)。精密量取一定量的P/GNPs,置于透析袋中(截留相对分子质量300 000),然后放置于100 mL 不同pH 值的PBS 溶液中(含有0.5%聚山梨酯80,pH 7.4、6.5、5.0),在37 ℃、200 r/min 的条件下恒温振荡。分别于0.5、1、2、4、8、12、24 h 取样1 mL,并补充相同体积的新鲜释放介质,按“2.3.1”项下测定释放介质中药物的质量浓度,以释放时间为横坐标、累积释放率为纵坐标绘制释放曲线,结果见图3。P/G-NPs 的药物释放未见明显突释。对于紫杉醇,在不同pH 值释放介质中均释放快于GMP,尤其在pH 7.4 条件下24 h累积释放率达到50%。对于GMP,随着释放介质pH 值的降低,P/G-NPs 释放速度逐渐加快,pH 值为5 介质中在24 h 药物累积释放率达到34%,分别是pH 7.4、6.5 条件下释放度的3.8、1.7 倍,说明P/G-NPs 中GMP 的释放具有一定的pH 值敏感性,对肿瘤内部酸性环境具有响应性。

图3 P/G-NPs 中的GMP 和紫杉醇在不同pH 值介质中的体外释放曲线 (n = 3)Fig.3 In vitro release profiles of GMP and PTX from P/G-NPs in media with different pH values (n = 3)
2.5 药物动力学研究
2.5.1 血浆样品处理 取大鼠眼眶静脉血300 μL加入到肝素钠抗凝血管中,8000 r/min 离心10 min,取上清液备用。取血浆100 μL,精密加入质量浓度为1.0 μg/mL DOX 的甲醇溶液50 μL 作为紫杉醇的内标,加入质量浓度为0.1 μg/mL 头孢克洛内标水溶液50 μL 作为GMP 的内标,涡旋5 min,再加入300 μL 甲醇沉淀剂,继续涡旋5 min,10 000 r/min离心5 min,取上清液按照“2.3.1”项下方法进样检测。
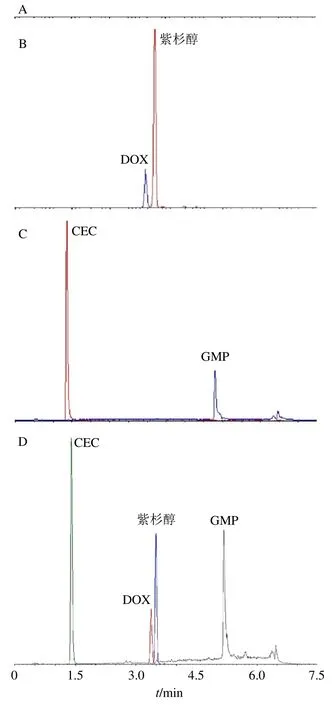
图4 空白血浆 (A)、空白血浆加紫杉醇和DOX (B)、空白血浆加GMP 和CEC (C)、空白血浆加紫杉醇、DOX、GMP和CEC (D)的HPLC 图Fig.4 HPLC of blank plasma (A),plasma spiked with PTX and DOX (B),plasma spiked with GMP and CEC (C),and plasma spiked with PTX,DOX,GMP and CEC (D)
2.5.2 专属性考察 专属性考察结果如图4 所示,在此色谱条件下,可实现同时检测溶解度不同的紫杉醇和GMP,与血浆内源性物质分离良好。
2.5.3 精密度实验 精密称取紫杉醇和GMP 对照品适量,用甲醇溶解并定容,摇匀,再次稀释至系列质量浓度,得到混合对照品溶液,将空白血浆与对照品溶液混匀,按照“2.5.1”项下操作,得到紫杉醇的质量浓度分别为0.1、2.0、20.0 µg/mL,GMP的质量浓度分别为0.05、1.00、10.00 µg/mL 的低、中、高3 个质量浓度的样品,平行制备5 份样本进行检测。每个质量浓度在同1 d 内检测3 次,分别计算紫杉醇和GMP 的日内精密度;相同的方法下每个浓度连续检测3 d,分别计算紫杉醇和GMP 的日间精密度,结果见表3,结果显示该方法日内、日间精密度RSD<15%,符合方法学要求。

表3 紫杉醇和GMP 血浆样品的精密度考察 (n = 5)Table 3 Precision of PTX and GMP plasma samples (n = 5)
2.5.4 提取回收率 配制质量浓度分别为0.1、2.0、20.0 µg/mL 的低、中、高3 个质量浓度的紫杉醇血浆样品,质量浓度分别为0.05、1.00、10.00 µg/mL的低、中、高3 个质量浓度的GMP 血浆样品,按照“2.5.1”项下方法分别测定紫杉醇、GMP 的峰面积(A实测)。
精密称取紫杉醇和GMP 对照品适量,用甲醇溶解并定容,摇匀,再次稀释至系列浓度,得到混合对照品溶液,将空白血浆与对照品溶液混匀,按照“2.5.1”项下操作,得到紫杉醇的浓度分别为0.1、2.0、20.0 µg/mL、GMP 的浓度分别为0.05、1.00、10.00 µg/mL 的低、中、高3 个质量浓度的样品,测定紫杉醇和GMP 的峰面积(A理论)。计算紫杉醇和GMP 的提取回收率(提取回收率=A实测/A理论),平行5 次。结果表明,高、中、低3 个质量浓度的紫杉醇提取回收率均在86.4%~95.5%,高、中、低3个质量浓度的GMP 提取回收率均在 80.3%~94.4%,符合生物样品检测要求。
2.5.5 药动学实验 SD 大鼠12 只,体质量(200±20)g,随机分为2 组,实验前禁食不禁水。按照16 mg/kg GMP 和2.0 mg/kg 紫杉醇的剂量尾iv 分别注射P/G-Free(按照规定剂量将GMP 溶解于紫杉醇注射液中)、P/G-NPs,分别于给药后1、4、7、15、30 min 及1、2、4、8、12 h,大鼠眼眶静脉丛取血0.5 mL,放置于低温离心机6000 r/min 离心5 min,取上清按照“2.5.1”项下方法处理,检测紫杉醇及GMP 的血药浓度,数据经PK solver 软件处理,血药浓度-时间曲线如图5 所示,药动学参数结果见表4。
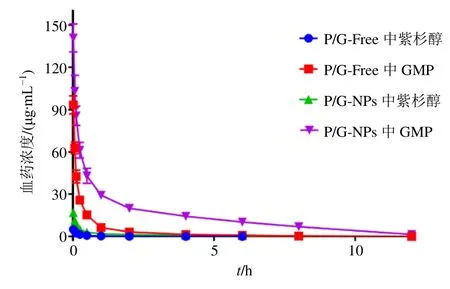
图5 静脉注射含有紫杉醇、GMP 不同制剂的血药-浓度时间曲线 (n = 6)Fig.5 Plasma concentration-time curves of PTX and GMP after iv injection of different preparations (n = 6)
3 讨论
多成分药物联合治疗在肿瘤等疾病领域愈发重要,将多种成分同时包载于纳米载体中,可以保持药物活性的同时,进一步发挥其相加作用而增强制剂疗效[19]。本研究采用核-壳结构纳米粒对紫杉醇、GMP 进行联合包载。紫杉醇、GMP 分别从抑制微管蛋白解聚、阻止DNA 聚合酶链延长、降低核苷酸还原酶活力等方面对肿瘤细胞起到杀伤作用[20]。紫杉醇/吉西他滨联合应用与单纯紫杉醇的III 期试验表明,在紫杉醇中加入吉西他滨,对先前接受蒽环类药物治疗的乳腺癌妇女,是一种有效的治疗方法[21]。本实验设计金正均Q值法实验也证明了紫杉醇及GMP 单药在物质的量比为17∶1 的条件下可以对降低MCF-7 细胞细胞活力起到协同作用。
本研究采用反相微乳法制备了粒径可控的GMP-Cores,并用薄膜分散法在GMP-Cores 表面包裹非对称的磷脂双分子层,并将脂溶性的紫杉醇嵌入于磷脂双分子层,双分子层修饰cRGD 靶向长循环磷脂,制备得到主动靶向P/G-NPs。体外释放结果显示,和GMP 相比,紫杉醇释放速度更快,原因在于紫杉醇位于磷脂双分子层疏水区,而GMP包裹于内部核心颗粒中,因此紫杉醇更容易接触释放介质。由于GMP 是在发生磷酸盐与钙盐共沉淀的过程中,包裹于Ca-P 颗粒中,其释放特性具有一定的酸敏感特性,体外不同pH 释放介质对GMP 的体外释放行为的影响也显示,Ca-P 在接近内涵体酸性pH 条件下(pH 5.0~5.5)可快速溶解,GMP 释放速度快于pH 7.4、6.5,这有利于实现纳米粒在内涵体内造成渗透压的失衡致内涵体破裂,释放药物进入胞浆中,使其具有自发的胞浆释药特性。

表4 P/G-Free 和P/G-NPs 中紫杉醇和GMP 的药动学参数 (n = 6)Table 4 Pharmacokinetic parameters of PTX and GMP in P/G-Free and P/G-NPs (n = 6)
药动学结果显示,P/G-NPs 中紫杉醇和GMP的最大血浆浓度(Cmax)均显著高于P/G-Free。P/G-NPs(AUC0~t)的面积比P/G-Free 高6 倍,平均停留时间(MRT0~t)增加2.3 倍。与P/G-Free 相比,P/G-NPs 中紫杉醇和GMP 的Vz值明显降低,分别为50.3%和72.1%,说明与P/G-Free 相比,P/G-NPs 可实现较好的长循环效果,这与纳米粒中PEG2000 修饰磷脂相关,PEG 可在纳米粒表面水化形成屏障[22],可减少血液中蛋白对其的吸附,还可帮助纳米粒逃避网状内皮吞噬系统的捕获,使得纳米粒在血液系统中的稳定性明显提高。
在长循环PEG 链可延长药物在体内的循环时间的同时,P/G-NPs 表明修饰 DSPE-PEG2000-cRGD,其靶向基团cRGD 可与癌细胞表面αvβ3/β5整合素受体特异性结合[23-24],进而通过内吞作用进入肿瘤细胞,有望进一步增强纳米粒靶向性,在肿瘤部位发挥药物协同作用,提高药物抗肿瘤效果。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
现代临床医学(2022年4期)2022-09-29
昆明医科大学学报(2021年4期)2021-07-23
中成药(2019年12期)2020-01-04
中成药(2018年7期)2018-08-04
中成药(2017年12期)2018-01-19
实用口腔医学杂志(2017年6期)2017-09-19
中成药(2017年5期)2017-06-13
中外医疗(2016年15期)2016-12-01
海南医学(2016年8期)2016-06-08
哈尔滨医药(2015年2期)2015-12-01