
介孔Cu/MS-1 催化羟基化苯酚合成苯二酚
2021-02-02 11:27余天华黄启朋司徒成
无机盐工业 2021年2期
余天华,黄启朋,司徒成
(贵州师范大学生命科学学院,贵州贵阳550001)
苯二酚是一种重要的工业产品和原材料, 广泛用于医药、染料和橡胶等精细化学品工业[1-3]。 苯胺氧化法和二异丙苯过氧化法是早期工业制备苯二酚的主要方法,但存在其步骤多、原子利用率低和对环境不友好等缺点, 不符合化学工业绿色化发展的趋势[1-2]。 因此,开发了一种简单的苯酚一步羟基化合成苯二酚的方法,且迅速成为研究的焦点,如苯酚的气相、液相中的均相和多相催化羟基化等反应[1,4-7]。但气相反应伴有能耗高、产生COx和结焦等现象[4-5];液相均相催化反应则存在产物与催化剂不易分离,催化剂难重复利用等不足[1,6];而液相中的多相催化反应具有反应条件温和、催化剂可重复利用、原子利用率高等优点,倍受研究者的青睐。
1975 年,Ube Industries,Lt. 首次报道以双氧水为氧化剂、直接羟基化苯酚、一步制备苯二酚时,铁和铜基催化剂对该反应表现出良好的催化活性。 由于铁和铜易得,价格低廉,受到学者的广泛关注[7-8]。而液相反应中,铁基催化剂不稳定,催化剂中的活性铁物种易流失,重复利用性差[3]。 铜基催化剂中的铜物种则易与载体相互作用, 形成稳定的催化活性中心[9]。 目前,铜基催化剂的研究主要包括含铜纳米粒子、含铜无机盐、嵌入及负载型的铜基催化剂,特别是以各类分子筛为基础, 通过各种手段引入铜的催化剂,由于制备简单、制作成本低、稳定性强和对环境友好等特点,吸引了许多研究者的兴趣[9-11]。Cu-TS-1[9]、Cu-MCM-41[12]、Cu-SBA-15[13]和Cu-CMM[14]都已被用于苯酚的直接羟基化,而且具有较好的活性。这些催化剂在制备时,将分子筛的合成和活性成分的引入合并,简化了步骤,但很难控制硅盐的水解和进入催化剂的活性组分的含量, 进入骨架的活性原子也会降低催化剂的稳定性。 而且上述稳定性强的催化剂孔径小, 不利于反应物及生成物分子在孔中的扩散; 孔径大的催化剂其孔壁为无定型结构,稳定性差,在伴有搅拌的液相反应中容易破碎, 从而导致活性物种的大量流失, 难以重复利用,因此限制了其在工业领域的广泛应用[1]。
负载型铜基催化剂的制备包括载体的合成和活性成分的加入两个步骤,可选择和合成载体,调控活性中心在载体上的分布, 使其具有更优越的催化活性和重复利用性。基于前期工作[15],采用溶胶凝胶法制备具有MFI 结构的S-1 时,加入十六烷基三甲基溴化铵(CTAB)模板剂,获得了同时具有介孔和MFI结构的MS-1,以浸渍法引入铜物种,用系列表征方法对样品的结构进行分析,并使用正交实验法对反应的条件进行优化, 探究载体结构与催化活性物种形成及催化活性间的关系,为大分子液相反应催化剂的制备及选用提供相关理论和实践依据。
1 实验部分
1.1 原料与试剂
正硅酸乙酯(TEOS)、NaOH、异丙醇(IPA)、CTAB、Cu(NO3)2·6H2O、四丙基氢氧化铵(TPAOH),均为分析纯,使用前未进行任何纯化处理。
1.2 样品制备
取45.0 g TEOS 置于500 mL 的烧杯中,室温搅拌下滴加40.0 g IPA,保持溶液透明条件下,依次滴加80.0 g TPAOH(质量分数为20%)水溶液和80.0 g超纯水,先升温至328 K 水解1 h,继续升温至350 K水解8 h,过程中持续补充水,使溶液体积基本不变,冷却至室温,强烈搅拌下加入10.0 mL CTAB 的IPA溶液,并保持搅拌8 h。所得溶液转高压反应釜中,充入 0.1 MPa 的氮气,448 K 下晶化 7 d[15]。 过滤悬浮液,洗涤固体至中性,干燥、焙烧(以5 K/min 速率从室温升至373 K 保持0.5 h; 再以5 K/min 的速率升到 823 K,保持 10 h),获得 MS-1。
取一定量的MS-1,抽真空至约2.7 kPa 并保持0.5 h,加入等体积的 Cu(NO3)2水溶液,室温下静置24h,干燥后,在823K 下焙烧10h。获得系列Cu/MS-1。
1.3 样品表征
采用LTD DX-1000 CSC 射线衍射仪分析样品的晶体结构。 用Trisar3020 型比表面积测试仪分析样品比表面积、 孔体积及孔径等结构参数。 用TU1901 型紫外光谱仪、Ultra DLD(KRATOS)型 X 射线电子能谱和实验室组装的H2的程序升温还原分析仪分析样品中的铜物种。
1.4 样品催化活性评价
依次取15.0 mL 超纯水、1.0 g 苯酚及一定量的催化剂置于50 mL 的两颈圆底烧瓶中, 加热至设置温度,在搅拌、回流条件下,用蠕动泵以0.1mL/min 加入H2O2,反应一段时间后,冷却至室温,过滤,洗涤并定容。 用配有Waters 2487 紫外检测器和C-18 柱的Waters1525P 高效液相色谱仪进行定性及定量分析。
反应后,经过滤、洗涤获得的催化剂,在373 K下烘干, 按照上述反应和检测条件考察催化剂的稳定性。
2 结果与讨论
2.1 样品XRD 表征
图 1 为样品的 XRD 谱图。 由图 1 可见,7~9°、23~25°和 45°处均有强的 MFI 结构的特征衍射峰,表明CTAB 的加入没有明显影响MS-1 的晶化度,样品具有高晶化的 MFI 结构[15-17]。
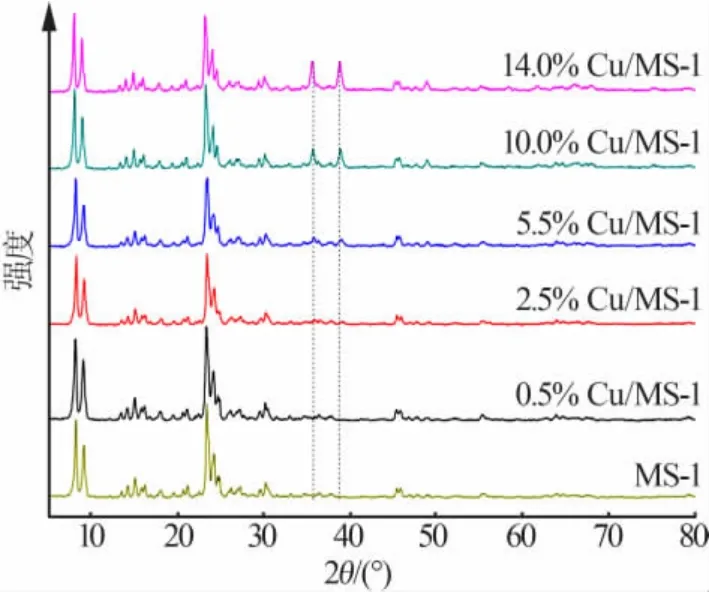
图1 样品的XRD 谱图
引进Cu 物种后,MFI 的特征衍射峰的峰位和强度基本没发生变化。 Cu 质量分数在2.5%以下的样品,XRD 谱图没有新衍射峰生成;Cu 质量分数为5.5%时,36.5°和 38.8°处出现 2 个弱的衍射峰,且峰强度随铜含量增加而增强;所有含Cu 的样品,43.2、50.4、74.1°处均没发现衍射峰。 上述结果说明,铜的引入没有影响样品的MFI 结构,当Cu 的负载量大于5.5%后,CuO 物种开始聚集,形成大颗粒的CuO,样品中没有 Cu2O 存在[18]。
2.2 样品N2 的等温吸附-脱附分析
图2 为样品的N2的等温吸附-脱附和孔径分布曲线,表1 为样品的结构性质。 由图2 可见,所有样品的N2的等温吸附-脱附曲线中,相对压力大于0.9后出现了明显的吸附现象, 孔径分布曲线中孔径主要集中在 2~4 nm,平均孔径为 2.7 nm 左右(表 1),小于2 nm 处存在少量的微孔。 表明CTAB 加入后,具有 MFI 结构和介孔的 MS-1 已被合成[19-20]。

图2 样品的N2 的等温吸附-脱附(A)和孔径分布曲线(B)

表1 样品的结构性质
引入Cu 物种后,随着铜含量的增加,平均孔径略增大,N2的等温吸附-脱附曲线的滞后环、表面积和孔体积均有减小的趋势。 这是由于随着Cu 含量的增加,更多的Cu 物种进入孔道且附在孔壁上,从而占据孔的空间,使样品的比表面积和孔体积减小;另一方面则是由于大量Cu 的引入导致Cu 物种形成大的颗粒,甚至烧结,从而堵塞了样品中一些小的孔口而导致比表面积和孔体积减小,平均孔径增大[21]。从孔径分布的曲线可以看出,随着铜含量的增加,微孔基本消失,所有样品均属于介孔材料。
2.3 样品DR UV-vis 光谱表征
图3A 为样品的DR UV-vis 谱图。从图3A 可以看出,MS-1 在 200~800 nm 没有吸收峰。 引入 Cu 物种后,吸收峰出现,随着Cu 含量的变化,吸收峰的强度发生有规律的变化。文献将230、440、600~800 nm处的吸收峰分别归属于 Cu—O—Si、Cu—O—Cu 和高分散度的 CuO 物种[14-22]。
为识别铜物种, 用Peakfit v4.12 软件对样品的UV-vis 谱图进行处理,结果如图 3B 和 3C 所示。 由图3B、3C 可知 , 当Cu 质量分数在2.5%以下时,有Cu—O—Si 和高分散度的CuO;Cu 质量分数达5.5%时,开始出现 Cu—O—Cu 的物种。 样品中 Cu—O—Si、Cu—O—Cu 和高 分散 CuO 峰 面积 比 值 (S230∶S440∶S600~800),从 w(Cu)=0.5% 时的 1.0/0.8/3.0 变为 w(Cu)=14.0%的1.0/3.0/6.0。 值得注意的是,随着Cu—O—Cu物种的出现及相对含量的增加,Cu—O—Si 物种的相对含量有减小的趋势。
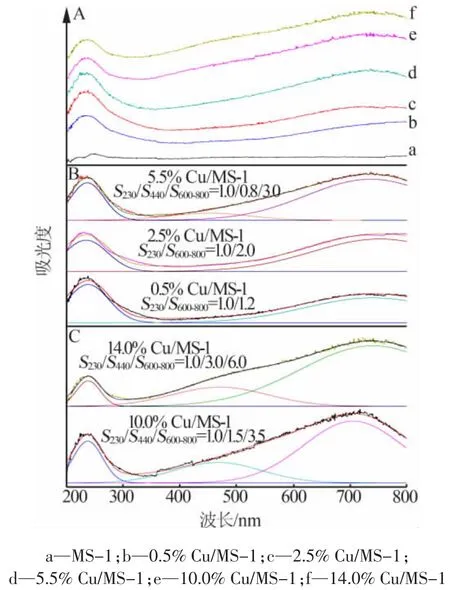
图3 样品的DR UV-vis 谱图
结果表明,Cu 含量较低时,Cu 易进入 MS-1 氧缺陷位或与其表面的羟基结合,形成骨架内的Cu—O—Si 物种, 当氧缺陷位大部分被Cu 占据后,剩余的Cu 能很好地在MS-1 的表面及孔中进行扩散和分布,形成高分散度的CuO 物种;而Cu 含量太高,焙烧时Cu 物种易烧结,形成Cu—O—Cu 物种。
2.4 样品XPS 表征
图 4 为样品的 XPS 谱图。 谱图中结合能约934.0 eV 的宽峰是CuO 负载于氧化物载体的特征峰,结合能在942.0~944.0 eV 处、强度稍弱的峰为卫星峰,说明样品中Cu 物种是以Cu2+的形式存在。 所有峰的峰强度随着Cu 含量的增加而增强, 拟合Cu 2p能谱图, 在电子结合能为933.0 eV 和约934.5 eV处有2 个峰,934.5 eV 处的峰随Cu 含量增加,电子结合能有逐渐增大的趋势, 这是由于Cu 含量增加后,铜物种发生聚集,且随铜含量增大,聚集也更加严重[3,18,23]。 结果与 XRD 和 DR UV-vis 结果相吻合。

图4 样品的XPS 谱图
2.5 样品H2-TPR 表征
图5 为样品的H2-TPR 曲线。由图5 可见,550 K处的尖峰、550 K 和650 K 处出现的2 个峰和700 K以上的峰分别为:1)Cu2+一步还原为Cu0的耗氢峰;2)Cu2+先还原为 Cu+, 再还原为 Cu0的耗氢峰;3)烧结的Cu 物种或Cu 物种与载体发生强的相互作用生成难还原的硅酸盐还原时的耗氢峰[24-25]。 当铜含量增加时,550 K 处的尖峰在曲线中峰面积占比先升后降, 在5.5%Cu/MS-1 中其比例最大。 表明Cu物种主要以一步还原的Cu2+存在, 继续增加铜的含量,二步还原Cu2+占比增加,且难还原的Cu2+占比急剧增加。 结果与 XPS 和 DR UV-vis 结果一致。 说明样品中3 种不同形式存在的Cu2+与引入Cu 的含量有关,含量较低或过高时,易形成难还原的Cu2+和经二步还原的Cu2+。

图5 样品的H2-TPR 曲线
2.6 样品催化性能的考察
采用正交实验法 L25(56)对 Cu 基催化剂存在下、 双氧水直接氧化羟基化苯酚的实验条件进行优化[26],以反应时间、催化剂种类、催化剂用量、反应温度、双氧水与苯酚的物质的量比、双氧水浓度为6 因素,对每个因素各取5 水平(表2 为正交实验设计表)进行实验,正交实验结果见表3。
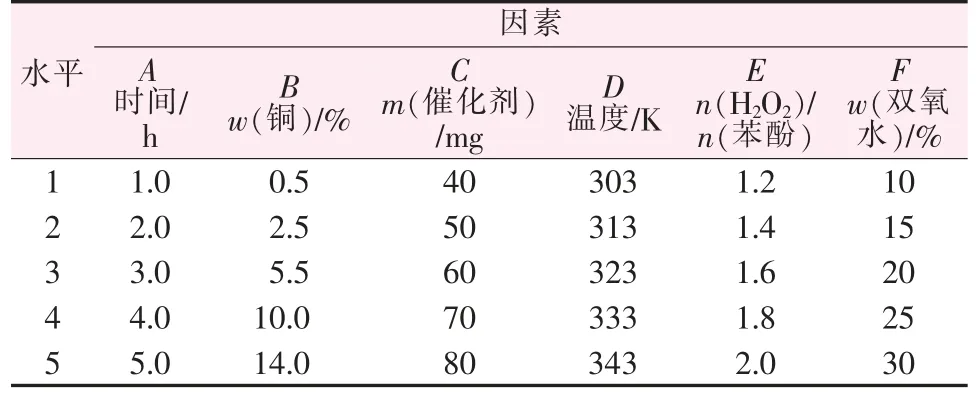
表2 正交实验设计
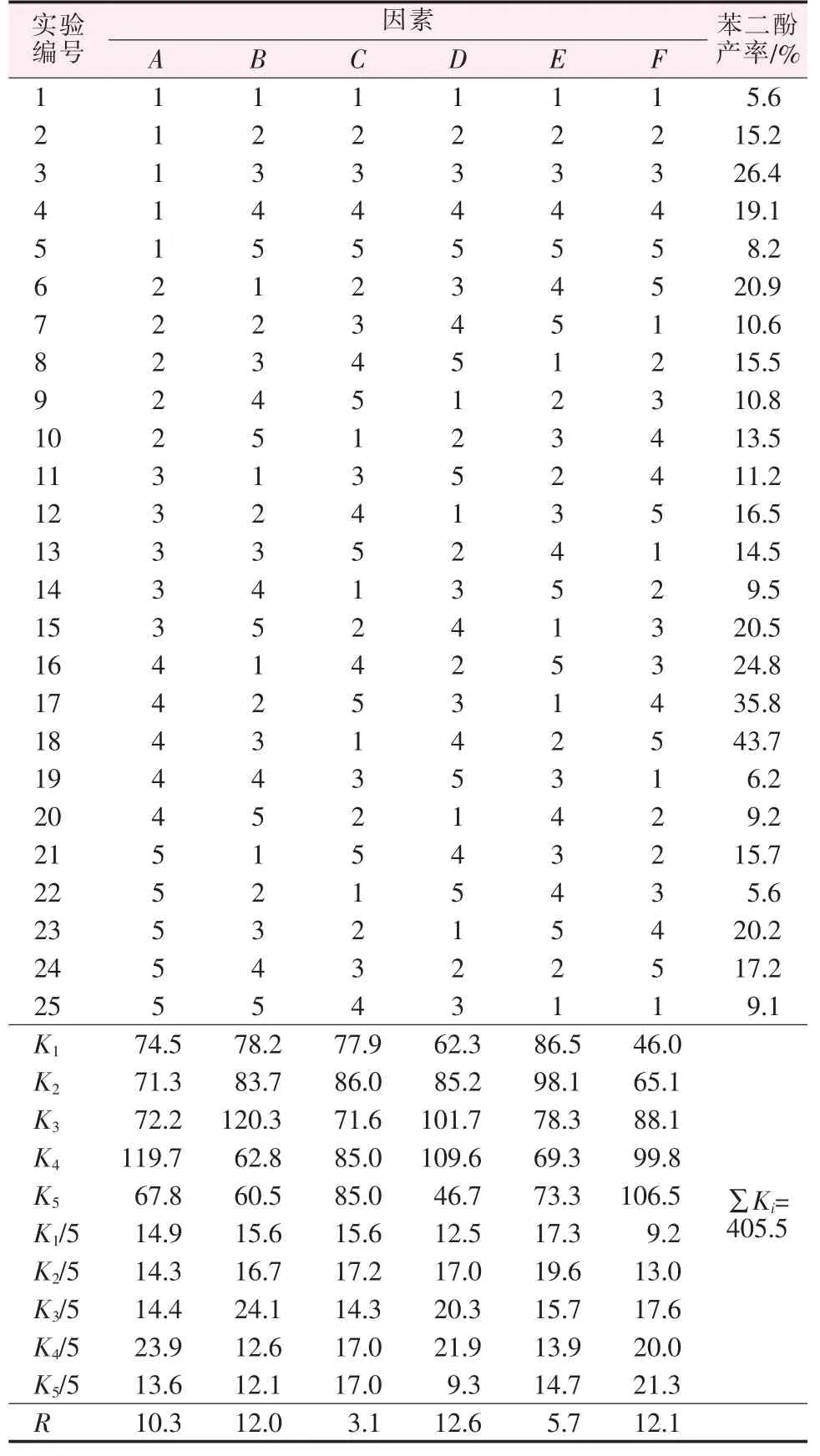
表3 苯酚直接羟基化生成苯二酚实验条件优化结果
通过正交实验法得到优化方案为A4B3C2D4E2F5,即反应时间为4 h、 催化剂中Cu 质量分数为5.5%、催化剂用量为 50.0 mg、反应温度为 333 K、n(H2O2)/n(苯酚)=1.4、双氧水质量分数为30%。 所做的实验中没有包括优化获得的结果。在优化条件下,进行目标反应,苯二酚的产率为47.2%,选择性为86.7%。高于优化过程中获得的最优产率(43.7%),说明所得方案为最优方案。目标反应中,反应因素影响从大到小顺序为反应温度、双氧水浓度和催化剂种类、反应时间、n(H2O2)/n(苯酚)、催化剂的用量。 当温度升高时,有利于反应的进行,但当温度高于333 K 时,双氧水的分解加剧,导致产率下降;反应时间延长和n(H2O2)/n(苯酚)增大,产物量会增加,但反应时间过长或 n(H2O2)/n(苯酚)过大,会导致生成的苯二酚深度氧化,形成过氧化产物;催化剂用量增加,可以提供更多的活性中心,有利于反应进行,但过多催化剂将促进H2O2分解为O2,导致H2O2有效浓度降低,从而降低产物的收率[14];双氧水的浓度有利于反应进行,考虑实验的安全,采用常用的质量分数30%为佳。
活性考察中, 催化剂中Cu 含量对催化活性的影响显著。 在上述优化条件下,以MS-1 为催化剂,没有检测到苯二酚, 表明铜物种是目标反应的催化活性物种。 为了考察催化剂中不同Cu 物种对目标反应的催化活性,在优化条件下,对铜含量不同的样品进行实验,其结果如图6 所示。 由图6 可见,随着铜含量升高, 苯二酚产率和苯酚的转化率均先升后降,在铜质量分数为5.5%时达到最高;苯二酚的选择性在铜质量分数为5.5%前基本保持不变, 大于5.5%后有明显降低的趋势。 通过XPS、DR UV-vis和H2-TPR 对样品的分析可知,样品中Cu 物种的存在形式包括高分散 CuO、Cu—O—Si 和 Cu2O2。 3 种铜物种绝对含量均随铜含量的增加而增加,相对含量中Cu—O—Si 随铜含量的增加而减少,高分散CuO 和Cu2O2随铜含量的增加而增加。 其中5.5%Cu/MS-1 中高分散、易被还原的CuO 物种相对含量的比例是最大的。 根据文献报道[3,8,12-14],采用双氧水为氧化剂,Cu 基催化剂催化羟基化苯酚制备苯二酚是自由基反应,主要催化活性中心是高分散易还原的CuO 物种。 所有催化剂中,5.5%Cu/MS-1 催化活性最好,尽管含铜5.5%以上的催化剂中CuO 物种的绝对数量也较多,但由于其焙烧形成Cu2O2物种较多,引起催化剂表面积下降,而且Cu2O2会促进H2O2的分解,导致催化活性降低。 结果与文献报道一致。
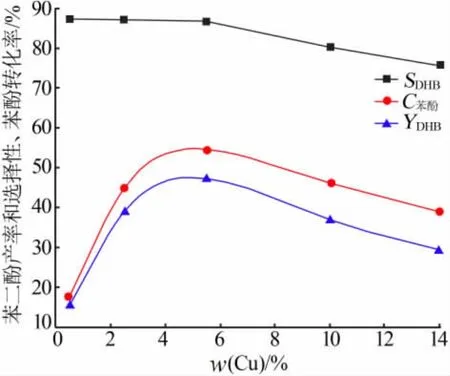
图6 催化剂中Cu 含量对苯二酚的产率和选择性及苯酚转化率的影响
实验还考察了催化剂结构对目标反应催化活性的影响,以5.5%Cu/S-1 为催化剂,在上述优化条件下进行反应, 苯酚的产率和选择性分别为24.5%和75.4%,远低于同样条件下的5.5%Cu/MS-1。 说明具有介孔的Cu/MS-1 更有利于大分子反应物和产物在孔道中的扩散,从而提高产物的产率及选择性。
在优化条件下, 考察了活性最高的催化剂的稳定性,结果见表 4。 由表 4 可见,重复使用 5 次后,催化剂中铜的含量和产物的产量均有减少, 但与新鲜催化剂相比变化不大,而苯二酚的选择性基本不变。表明载体高晶化、强稳定性的MFI 结构和铜物种与载体间的相互作用,使催化剂具有强的稳定性,可重复使用。
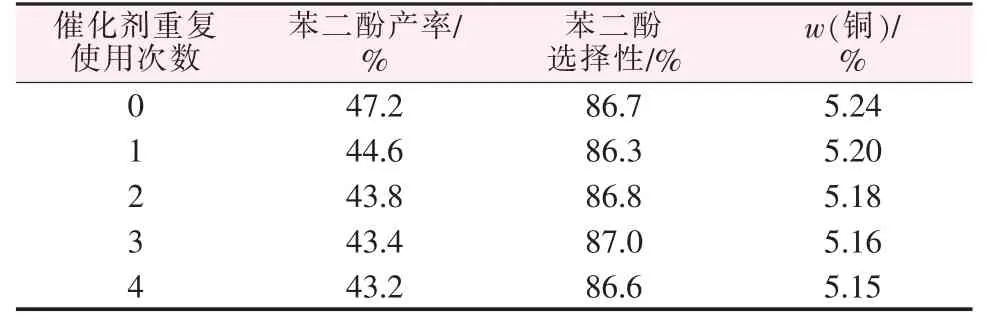
表4 催化剂稳定性的考察
3 结论
采用溶胶凝胶法制备S-1 时, 加入CTAB 为模板剂,获得了具有介孔结构的MS-1。 在MS-1 中引入铜物种时,铜物种与载体作用,生成高分散CuO、Cu—O—Si 和 Cu2O2,其中高分散 CuO 易于还原,是目标反应的主要活性物种。 正交实验法优化结果发现,5.5%Cu/MS-1 催化活性最高,在最佳条件下,苯二酚的产率和选择性分别为47.2%和86.7%。与5.5% Cu/S-1 相比,具有介孔结构的5.5% Cu/MS-1 具有更优越的活性,且其稳定性强,能够重复使用。
猜你喜欢
城市道桥与防洪(2022年3期)2022-05-08
能源化工(2021年6期)2021-12-30
云南化工(2020年11期)2021-01-14
云南化工(2020年11期)2021-01-14
云南化工(2020年11期)2021-01-14
合成技术及应用(2021年1期)2021-01-07
装备维修技术(2020年5期)2020-11-20
应用化工(2020年9期)2020-09-29
矿产综合利用(2020年1期)2020-07-24
分析化学(2017年9期)2017-10-16
