
Ni/W 比对NiWCx 催化剂加氢性能的影响
2021-02-02 11:27:46张海永刘思琪刘龙吟
无机盐工业 2021年2期
张海永,张 璐,刘思琪,刘龙吟
[中国矿业大学(北京)化学与环境工程学院,北京100083]
自R.B.Levy 等[1]报道了碳化钨具有类似贵金属的电子结构和催化特性以来, 人们对过渡金属碳化物在不同催化反应中的应用展开了广泛的研究。 过渡金属碳化物通常以体积较小的碳原子填充过渡金属原子密堆积间隙的形式形成“间充性合金”,一般具有六方密堆结构(如 β-Mo2C、η-Mo3C2、WC)或面心立方结构(如 α-MoC1-x、W2C)。 一般而言,碳化程度越高,则其催化行为越类似贵金属催化剂[2]。 过渡金属碳化物催化存在诸多反应,如石油加氢处理、烷烃重整、费托合成、烃类异构、甲烷重整、电催化分解水等。 研究发现,碳化物在涉氢反应中表现优异,展现出良好的对氢分子的吸附与活化性能[3]。 通过对油品加氢处理的研究,人们证实了碳化钼、碳化钨等催化剂具有良好的加氢活性[4],如 Mo2C/Al2O3在柴油中噻吩深度脱硫和芳烃加氢方面比传统的硫化钼催化剂更佳,加氢脱硫(HDS)活性可达商业硫化钼催化剂的 1.5~2 倍,V.Schwartz 等[5]和 D.J.Sajkowski等[6]以Mo2C/Al2O3对煤基馏分油进行长周期加氢时发现,其HDS、加氢脱氮(HDN)和芳烃预加氢脱硫(HYD)活性为 MoS2/Al2O3或 NiMoS/Al2O3催化剂的3~5 倍不等, 且具有较好的抗硫中毒性。 H.A.Al-Megren 等[7]以 Co-Mo 与 Co(Ni)-W 碳化物对吡啶做催化加氢实验, 发现吡啶在Co-Mo 催化剂上140 h内保持了100%转化率,远高于工业CoMoS/Al2O3催化剂。目前,在碳化物催化剂的研究中对碳化钼的研究较多, 而对碳化钨尤其是对助剂存在下的双组分碳化钨催化剂的研究较少。 一般而言,W 对于芳烃和含氮化合物的加氢效果要好于Mo。中国作为煤化工大国,每年通过热解、气化和液化过程产生大量富含芳烃的煤基油品,且其中含有一定量的N、O 和S杂原子化合物,与传统的石油馏分有较大的差异。因此,采用中国储量相对丰富的W 对煤基粗油进行加氢处理具有重要的意义。
本研究以不同Ni/W 比(原子比,下同)的NiW/Al2O3碳化物催化剂对萘、模型油和从真实煤基油品中分离出的芳烃组分进行催化加氢处理, 考察助剂Ni 对碳化钨催化剂的促进作用,并与硫化钨催化剂做了对比,为煤基芳香油品的提质加工提供依据。
1 实验部分
1.1 催化剂制备
按照(NiO+WO3)含量占焙烧后催化剂的30%(质量分数),改变Ni/W 比,分别以硝酸镍和偏钨酸铵为前驱体通过二步等体积浸渍法制备出以375~850 μm γ-Al2O3负载的 NiW/γ-Al2O3催化剂。 经120 ℃干燥 10 h 后, 在马弗炉中以 5 ℃/min 加热至600 ℃焙烧4 h,得到不同Ni/W 比的催化剂,分别以Cat-x 表示,其中 x 为催化剂中的 Ni/W 原子比。 所得催化剂的组成如表1 所示。
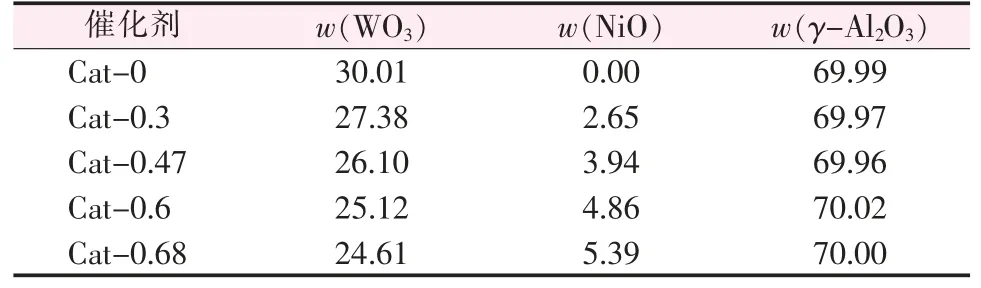
表1 不同Ni/W 原子比的NiW/γ-Al2O3 催化剂的组成 %
称取1 g 催化剂并以石英砂稀释10 倍后装填在固定床管式反应器中部恒温区。 反应前以甲烷氢气混合气(CH4与 H2体积比为 1∶4)通过程序升温碳化法对催化剂进行碳化[8],气体空速为 1 200 h-1,以10 ℃/min 的速率从室温快速升至 300 ℃,而后以1 ℃/min 的速率从 300 ℃升温至 700 ℃,在 700 ℃下保持3 h。在氩气环境中降温至反应温度进行加氢反应,或降至常温后以氧气和氩气混合气(O2与Ar 体积比为1∶99)进行钝化以做表征。
1.2 催化剂表征
催化剂孔结构采用ASAP 2460 型吸附仪测定。采用 Smartlab3000V 型 X 射线衍射仪对样品做XRD 表征。 采用TP-5100-Ⅱ型多功能吸附仪对催化剂进行程序升温还原(H2-TPR)。 以MERLIN Compact 型扫描电子显微镜做SEM 表征。
1.3 催化剂评价
实验以萘(质量分数为5%的正庚烷溶液)、模型油溶液(萘、苯酚、吡啶质量分数分别为5%、1%、0.1%,溶剂为正庚烷)作为煤焦油模型化合物进行加氢实验,液时空速(LHSV)为 2 h-1,氢油比(体积比,下同)为 600∶1,反应压力为 3 MPa,收集 1 h 的样品进行分析。 按本组开发的方法[9]以溶剂萃取法从煤焦油中分离出芳烃组分,配制成质量分数为5%的甲苯溶液后进行加氢反应,LHSV 为1 h-1,氢油比为 1 200∶1,反应压力为 5 MPa,反应温度为 300 ℃,收集第2 h 的产物进行分析。 使用SP3420 型气相色谱仪进行模型化合物的分析,使用瓦里安3800 型气质联用仪对低温煤焦油芳烃富集组分进行分析。
2 催化剂分析表征
2.1 催化剂孔结构分析
表 2 为不同 Ni/W 比的 NiW/γ-Al2O3催化剂和载体的孔结构参数,其孔分布如图1 所示。由表2 可知,载体负载金属组分之后,比表面积、孔容、平均孔径都明显下降,Cat-0 比表面积为98.65 m2/g, 降幅最为明显;而随着Ni 含量增加,催化剂比表面积与孔容逐渐回升,Ni/W 原子比为0.47 时达到最大,而后比表面积与孔容开始下降或基本持平, 平均孔径则逐渐下降,至Ni/W 原子比为0.47 后略有回升。由图1 可以看出,催化剂与载体的孔径分布基本相似,为单峰型分布,主要孔径分布在4~24 nm,最可几孔径为10 nm。 但催化剂在7~14 nm 时的孔道分布比载体要少,因此孔径分布曲线相对较扁。并且当载体负载活性金属后在6 nm 附近出现了一个不太明显的小肩峰, 结合表2 中催化剂平均孔径比载体整体偏小的结果, 说明活性金属的负载整体减小了孔道直径,降低了10 nm 左右孔道的比例,生成了一些6 nm 左右的孔道。
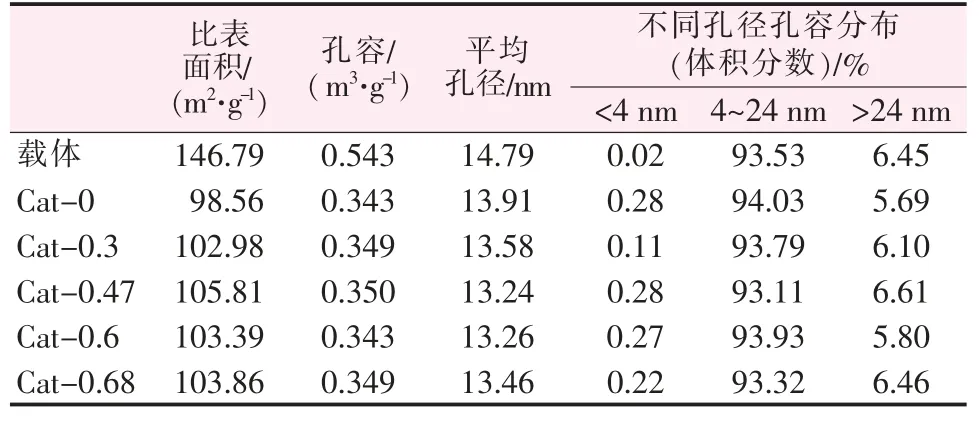
表2 不同Ni/W 原子比催化剂及载体的孔结构参数

图1 载体与Cat 系列氧化物前驱体孔径分布曲线
2.2 XRD 分析
图 2 为不同 Ni/W 比 NiW/γ-Al2O3催化剂的XRD 衍射谱图。 由图 2 可见,在 2θ 为 37.67、45.79、67.08°处 的 衍 射 峰 为 γ-Al2O3的 特 征 峰 ;23.61、24.37、28.64、33.30、40.49、54.83、62.31°处的衍射峰为WO3的特征峰。 一般认为,催化剂中WO3的单层分散阈值约为20%[10],而本研究所用催化剂中WO3的质量分数为24%~30%,在单独负载于氧化铝载体表面时(Cat-0)检测到明显的 WO3特征峰。 对比Cat-0.3 和Cat-0 的谱图可以看出,Ni 的加入使得WO3的特征峰明显降低, 尤其是 23.61、33.30°两个尖峰,此外 40.49、54.83°处的两个峰消失。 对比全部样品的谱线,23.61°和 28.64°处两个 WO3的特征峰随着Ni/W 比的增加呈现先降后增趋势,Ni/W 原子比从 0.3 增至0.47 和 0.6 时基本消失, 再增加至0.68 时其强度又回升, 说明WO3重新出现聚集现象。 此外,Ni/W 原子比增至 0.47 之后 23.61°处的强特征峰逐渐消失, 而在20.97°处出现非晶态的弥散峰, 其他氧化钨或氧化镍类的特征峰则减小甚至消失, 也印证了在此Ni/W 比范围内载体表面金属的分散性相对良好。根据文献报道,镍金属加入有助于抑制WO3在载体表面的聚集,能够明显改善催化剂表面金属的分散程度,但Ni 的促进作用存在阈值[11-13],从 XRD 谱图及孔结构表征可以看出,Ni/W原子比在0.47~0.6 时能较好地抑制晶体团聚。
将NiW/γ-Al2O3催化剂经程序升温碳化转化为碳化物,并经钝化后取出做XRD 测试,结果如图3所示。 由图 3 可见,在 37.84、39.36、61.60、74.70°处的衍射峰为 W2C(PDF 35-0776)的特征峰[14],43.29°处为 NiO(PDF 89-7131)。 碳化后催化剂的 XRD 谱线的变化趋势与氧化物相似,Ni 的加入使得W2C 衍射峰下降,Cat-0.3 与 Cat-0 中 W2C 在 37.84°处的特征峰与 γ-Al2O3的 37.67°的特征峰重合,但 Cat-0.47、Cat-0.6 和 Cat-0.68 中重合峰明显下降,61.60°处的衍射峰随Ni/W 比的增大也逐渐降低, 说明W2C 的分散性提高。 而随着 Ni 的增加,NiO 的衍射峰逐渐增强,这应归因于钝化过程中活性的Ni 金属被氧气氧化, 但并未检测到钨的氧化物种。 此外,XRD 还检测到非晶态的弥散峰及C 的峰,说明催化剂中存在大量分散均匀未形成大晶粒的组分,并且存在一定量的积炭。 根据孙军等[15]的研究,WO3先还原为 WO2,600 ℃WO2经碳化反应生成 W2C,且随温度升高碳化程度增加,680 ℃时WO2完全转化为W2C,温度升至 850 ℃时,W2C 完全转化为 WC 并且碳源气体在表面生成大量积炭。
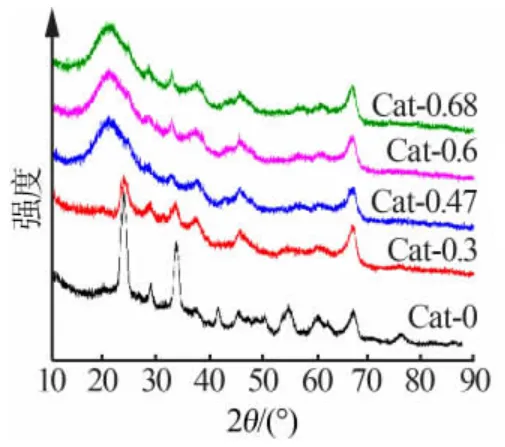
图2 NiW/γ-Al2O3 催化剂的XRD 谱图
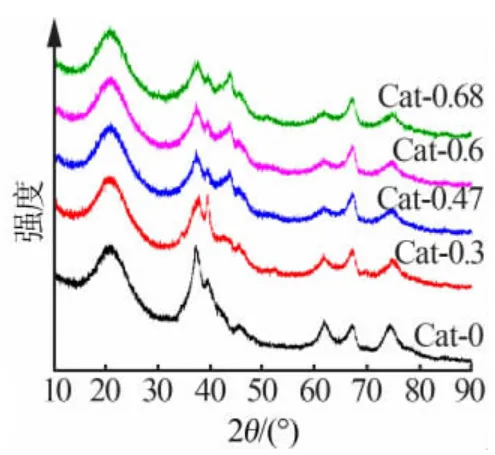
图 3 NiW/γ-Al2O3 催化剂碳化后的XRD 谱图
2.3 H2-TPR 分析
图 4 为不同 Ni/W 比的 NiW/γ-Al2O3催化剂的H2-TPR 谱图,由图4 可知,还原峰温及峰型可反映金属还原性的难易及均匀程度。 单组分WO3/γ-Al2O2催化剂Cat-0 的TPR 曲线只在820 ℃出现了WO3还原峰,随着Ni 的加入,于650 ℃左右出现NiO的还原峰,WO3的还原峰向低温迁移,另有一个860 ℃的还原峰应为与载体有强相互作用的WO3。 Ni/W原子比增至0.47 之后还原峰仅有650 ℃的主峰,并于700~800 ℃ 时 出 现 Ni-W-O 混 合 相 的 小 还 原 峰[14]。Ni 的加入使WO3的还原峰向低温移动,即其与载体间的相互作用减弱,使得这有利于WO3还原并碳化为W2C。 对比4 个含Ni 催化剂的还原曲线,Cat-0.3在650 ℃出现的宽峰应为NiO 和WO3两物种分别在640 ℃和 675 ℃的还原峰的重叠峰,Cat-0.47 中WO3在675 ℃的还原峰继续左移,两峰重合并且在650 ℃形成尖峰,说明当Ni/W 原子比增至0.47 之后NiO 和WO3在载体表面形成了大量相对均匀的物种,其还原性相比 WO3/γ-Al2O3提高很多。 在 730 ℃处出现的小肩峰应为Ni-W-O 晶相, 而860 ℃处与载体具有强相互作用的WO3的还原峰则基本消失。Cat-0.6 和Cat-0.68 与Cat-0.47 的还原曲线基本相似,但 Cat-0.6 中 650 ℃的还原峰较 Cat-0.47 高,而随Ni 含量的继续增加Cat-0.68 中此峰又略向左移至645 ℃。

图4 不同 Ni/W 比的 NiW/γ-Al2O3 催化剂的 H2-TPR 谱图
2.4 催化剂表面形貌
图5 为载体和催化剂的SEM 照片。 由图5 可以看出,负载金属组分后,催化剂表面的颗粒状结构增加,Cat-0.47 中颗粒状结构黏结于载体表面, 且颗粒大小与Cat-0 和Cat-0.3 相比有所降低,但Cat-0.68 中颗粒大小增加, 表面出现团聚现象。 此外,Cat-0.47、Cat-0.6 和Cat-0.68 中出现片状结构, 晶体分布相对更均匀。

图5 载体与催化剂的SEM 照片
3 催化剂性能评价
3.1 不同Ni/W 比NiW/γ-Al2O3 催化剂对萘的催化加氢性能
图6 为不同催化剂对萘加氢1 h 的反应效果。由图6 可见,萘加氢时经四氢萘生成十氢萘,图中6个催化剂对萘的加氢转化率相似, 均在97%以上,但深度加氢产物十氢萘的选择性却差异较大,随Ni/W 原子比的增大逐渐提高,至0.6 时达到最大,而后迅速下降, 其中Cat-0.47 与Cat-0.6 对十氢萘的选择性分别达到94%与98%,加氢深度较高,显示出非常好的加氢活性。说明Ni 的加入在一定范围内大幅促进了催化剂的加氢性能, 使得更多的加氢产物从四氢萘转化为十氢萘。
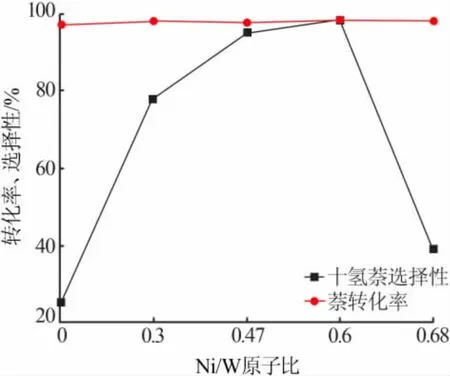
图6 不同催化剂对萘加氢的效果
3.2 温度对NiW/γ-Al2O3 催化剂催化萘加氢的影响
图 7 为 Cat-0.6 催化剂于 225~325 ℃下对萘的加氢效果。 由图7 可见,225 ℃时萘转化率就已达到98%,其十氢萘与四氢萘选择性分别为67%与30%。随着温度升高,十氢萘选择性逐渐增加,275 ℃时达到最大值98%,此后随温度的升高而降低,并且有裂化产物出现, 通过差减法得到300 ℃与325 ℃裂化选择性分别为25%与35%。 因载体氧化铝具有酸性,本研究中所制备的催化剂具有双功能催化剂的特点, 十氢萘在催化剂酸性作用下发生开环和裂化反应,降低了其选择性,所以实验选择相对适宜的温度为275 ℃。
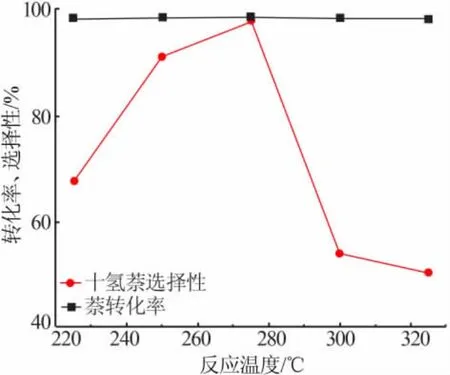
图7 温度对Cat-0.6 催化剂催化萘加氢的影响
3.3 NiW/γ-Al2O3 催化剂对模型油的加氢性能
图8 为模型油(萘为 5%、苯酚为 1%、吡啶为0.1%、溶剂为正庚烷)的催化加氢效果。 由图8 可见,随着Ni 含量的增加,萘的转化率先迅速增加后缓慢下降,与纯萘加氢时具有相似的趋势,体现出Ni 的添加对W 加氢活性的促进作用。 其中Ni/W 原子比为0.3~0.6 时萘的转化率相近,Ni/W 原子比为0.47 时达到最大值 85%, 与此同时,Cat-0.3、Cat-0.47 与Cat-0.6 对十氢萘的选择性也相近, 均在50%以上, 但Cat-0.68 对萘的转化率和十氢萘的选择性均降低,加氢活性下降。 此外,5 个催化剂对吡啶和苯酚的转化率表现出与萘相似的趋势, 即先增加后减小,但吡啶的转化率保持较高的水平,维持在85%~99%,而苯酚转化率从最低的Cat-0 的35%到最高的Cat-0.6 的88%出现明显波动。
与萘加氢结果相比, 吡啶与苯酚的加入使催化剂活性出现明显的降低, 表现较好的Cat-0.3、Cat-0.47 与Cat-0.6 催化剂对萘的转化率和十氢萘的选择性分别下降了15%和30%以上。而对比未添加Ni的Cat-0 催化剂对两种原料的加氢效果, 杂原子化合物的添加对萘转化率造成的抑制作用甚至可达90%以上。 因此,Ni 的加入可以提高催化剂抗杂原子能力,但Ni 对催化剂的促进作用存在阈值。
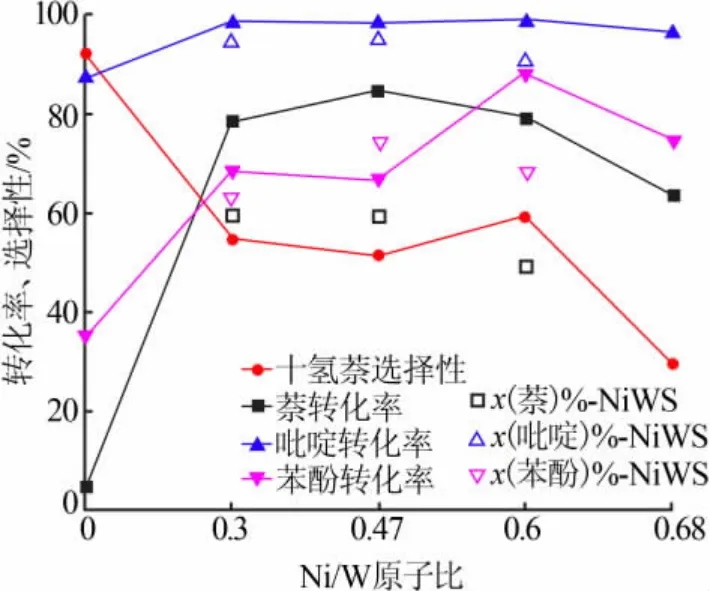
图8 不同催化剂对模型油催化加氢的效果
对硫化后的 Cat-0.3、Cat-0.47 和 Cat-0.6 这 3个催化剂同样做了模型油加氢性能评价, 与碳化物相比,其对萘的转化率整体要低20%以上,对吡啶的转化率略有下降,而对苯酚的转化率相近,各有优劣。但萘加氢产物中基本全部都为四氢萘,十氢萘的产率均低于0.3%。说明在本研究体系所涉及的催化加氢活性方面, 相同组成的NiW/γ-Al2O3催化剂其碳化物要比硫化物效果要更好。
3.4 NiW/γ-Al2O3 催化剂对低温煤焦油芳烃组分的加氢性能
通过上述研究, 选取加氢效果较好的Cat-0.3、Cat-0.47 和Cat-0.6 这3 个催化剂对低温煤焦油中萃取分离得到的芳烃组分做加氢处理,GC-MS 分析谱图如图9 所示。由图9 可见,原料中残留了作为萃取溶剂的NMP,经加氢后其含量明显降低,但仍相对较高。 经加氢后,重组分多环芳烃含量降低,而轻组分峰强度则增加。
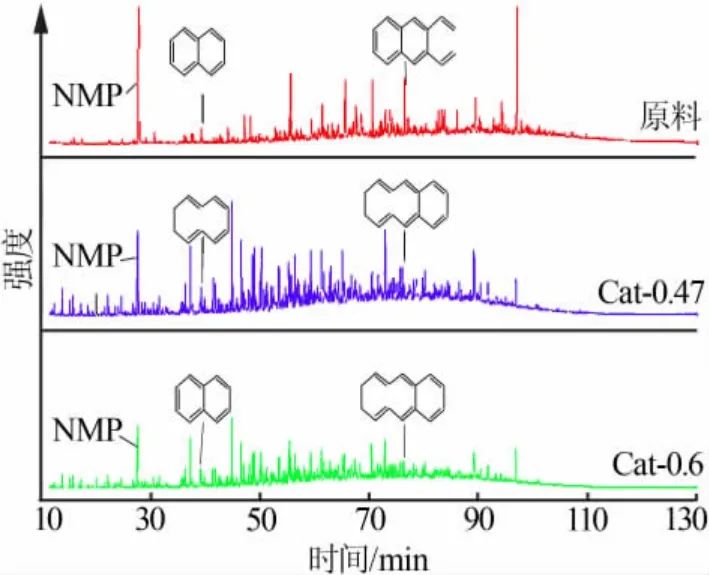
图9 不同催化剂对低温煤焦油芳烃组分加氢前后的GC-MS 谱图
扣除NMP 峰后通过简单面积归一法分析原料及加氢产物,各组分含量如图10 所示。 需要指出的是,因化合物种类众多,很难准确对各物质做精确定量,因此图中各组分的含量与实际组成有偏差,但作为粗略的横向对比可以大致了解不同样品中各组分的变化趋势。经分析,图10 中的原料主要为芳烃,占72.3%(质量分数,下同),杂原子化合物约为24%,环烷烃与氢化芳烃含量极少。 经过加氢饱和与加氢裂化反应, 产物逐渐轻质化, 杂原子化合物含量降低, 氢化芳烃与环烷烃含量明显增加, 经Cat-0.3、Cat-0.47 和Cat-0.6 处理后的产物中芳烃含量分别降至38.04%、19.19%和31.57%,且三环芳烃质量分数由30%左右降到6%以下,体现出良好的加氢效果。 3 种催化剂对杂原子化合物的脱除效果存在差异,分别下降了9.06%、18.36%和7.11%。
综合上述不同原料对催化剂性能评价的对比,Cat-0.6 与Cat-0.47 两个催化剂在萘和模型油的加氢实验中体现出相近的活性,杂原子化合物(苯酚和吡啶)的添加使Cat-0.47 与Cat-0.6 的差距缩小,在对煤焦油中芳烃组分加氢时,Cat-0.47 的加氢效果更是明显优于Cat-0.6,这与Ni、W 两种活性组分之间的相互作用和协同作用有关。 Ni 与W 均可活化氢分子并催化加氢反应,在油品加氢处理过程中,W和Mo 作为加氢主剂, 而Ni 和Co 则一般作为助剂成分,能够促进W(Mo)物种在载体表面的分散,降低其还原及硫化难度并更多地生成高活性的晶相。Ni 虽本身具有较好的加氢活性,但其抗杂原子能力不足;而WO3还原困难,不易生成加氢活性高的WCx物种。 因此,以 Ni 为助剂的 NiW 碳化物催化剂存在一个适宜的Ni/W 比, 这可以使催化剂的加氢活性达到最高, 并且能在一定程度上抵抗杂原子化合物的毒化作用。 从本研究可以看出,Ni/W 原子比为 0.47 时 NiW/γ-Al2O3碳化物催化剂对含有 N、O杂原子化合物的芳烃以及煤焦油中分离得到的芳烃组分具有更好的加氢活性。 NiW 硫化物催化剂对煤基油品加氢处理的相关研究也表明, 最佳Ni/W 比与本研究所得结论相近。

图10 不同催化剂对低温煤焦油芳烃组分加氢前后组成分析
4 结论
本研究采用二步等体积浸渍法制备了负载于γ-Al2O3载体上的不同Ni/W 原子比的NiW 催化剂,经程序升温碳化转化为碳化物后对芳烃模型化合物和煤焦油中分离得到的芳烃组分做了加氢处理,结合催化剂的理化表征结果, 得到结论:1)Ni 的加入可以降低γ-Al2O3载体表面WO3的团聚现象, 提高金属组分分散性, 并且可以减弱金属与载体的相互作用,提高金属氧化物的还原性,使其更易转化为金属碳化物。2)Ni 与W 均具有较好的加氢活性,但Ni的耐杂原子性较差, 因此芳烃加氢时Ni/W 比存在一个适宜的范围。 该系列NiW/γ-Al2O3碳化物催化剂对萘的加氢效果非常好,Ni/W 原子比为0.6 时最佳,适宜的温度为275 ℃。但添加苯酚和吡啶后的模型油加氢时Ni/W 原子比分别为0.47 和0.6 的性能相近, 煤焦油芳烃组分加氢时则Ni/W 原子比为0.47 的催化剂反应效果最佳, 芳烃质量分数下降53.1%。
猜你喜欢
上海金属(2022年6期)2022-11-25 12:24:20
云南化工(2021年6期)2021-12-21 07:31:00
环境卫生工程(2021年4期)2021-10-13 06:52:16
模具制造(2019年3期)2019-06-06 02:11:04
山东冶金(2018年6期)2019-01-28 08:14:50
上海金属(2016年4期)2016-11-23 05:38:50
橡胶工业(2015年2期)2015-07-29 08:29:44
水利建设与管理(2015年10期)2015-05-09 08:29:50
化工管理(2015年12期)2015-03-24 21:06:07
纯碱工业(2014年6期)2014-03-11 15:09:25
