
含能五唑阴离子及其衍生物的合成与性质研究进展
2021-01-26 03:02DominiqueWozniakDavinPiercey
工程 2020年9期
Dominique R. Wozniak, Davin G. Piercey
1. 引言
高能密度材料(high-energy-density material,HEDM)在炸药、推进剂和烟花中的广泛应用引起了研究人员的注意。不断增长的对更安全、高效、环保的HEDM的需求一直在影响着人类寻找这些独特材料。因此,无论是在实验上还是在计算上,人们已经对这类材料的合成和表征进行了很长时间的研究[1,2]。
“经典”炸药,如2,4,6-三硝基甲苯(2,4,6-trinitrotoluene,TNT)、环三亚甲基三硝胺(1,3,5-trinitro-1,3,5-triazacyclohexane, RDX)和1,3,5,7-四硝基-1,3,5,7-四氮杂环辛烷(1,3,5,7-tetranitro-1,3,5,7-tetraazacycloocatane, HMX)从同一分子中的燃料和氧化剂的组合中获得爆炸性能。2,4,6,8,10,12-六硝基-2,4,6,8,10,12-六氮杂四环十二烷(2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurzitane, CL-20)和八硝基立方烷(octanitrocubane, ONC)等“第二代”炸药,由于分子中存在笼状应力,因此比传统炸药具有更大的能量值[1–3]。
HEDM设计的最新动向之一是高氮和多氮化合物。所有高氮物质的能量值都来自形成延长的单键氮和双键氮体系的高热量。氮化学不同于碳化学;氮与碳不同,碳的键序增加会导致平均键能降低,而氮的键序增加则导致平均键能升高[4]。由于这种异常的特性,氮气中的单键和双键形成氮-氮三键,会释放大量能量。如此大的能量释放对于其在HEDM中的应用具有潜力,但也意味着多氮化合物是亚稳态的,从而使其合成和表征变得困难。在2016年之前,只有N5+和叠氮化合物阴离子被大规模合成[5,6]。
最近,五唑阴离子(cyclo-N5−)受到的关注显著增加。2016年之前,多项计算性研究准确地预测了cyclo-N5−的稳定性和存在性。对cyclo-N5−进行理论分析,使其在放热反应中分解为N3−和N2,同时释放出14.3 kcal·mol−1(1 kcal = 4.184 × 103J)的能量。该反应的势垒为27.2 kcal·mol−1[7,8]。生成热(ΔHf, 298 K)和电离势分别为59.6 kcal·mol−1和5.06 eV [8–10]。该数据表明在适当条件下解离cyclo-N5−的可能,同时也是许多合成化学家努力获得和了解这种阴离子的最初动机[11]。
Bazanov等[12]于2016年在四氢呋喃(tetrahydrofuran, THF )中对cyclo-N5−进行质谱检测,这是合成含cyclo-N5−化合物的几项重要进展中的第一步。在三年之内,大量的cyclo-N5−、其金属盐和其有机盐的合成全部实现[13–22]。Wang等[2]之前对理论和实验结果进行过广泛的综述。但是,由于该领域快速吸引了大量关注,因此为研究人员提供最准确和最新的信息至关重要。考虑到这一点,我们希望在本文中增加新的研究和解释,重点是已知五唑含能离子盐的合成和性质。在此,阐述了cyclo-N5−及其相关化合物合成背后的原理。
2.c yclo-N5−的前驱体
对于多氮物质,特别是cyclo-N5−的实验,人们已研究了20年[23]。即使付出了巨大的努力,cyclo-N5−的合成仍然只有两条经过验证的实验路线(图1)[2]。目前最流行的路线是取代芳基重氮盐与叠氮阴离子反应生成取代的五唑中间体,然后该中间体环化形成取代的五唑前驱体,可用于制备cyclo-N5−[24–26]。另一个较冷门的路线涉及从N3−和N2直接生成cyclo-N5−,但是该过程涉及金刚石砧座单元中高压和高温反应的极端条件,与第一个途径相比,本体合成的可行性较低。遗憾的是,芳基五唑(ArN5)前驱体的生产仍然是一项艰巨的任务。cyclo-N5−环的芳香度对其稳定性至关重要,它依赖于连接在芳基上的取代基。此外,有实验已经证明,从ArN5前驱体裂解cyclo-N5−而不破坏五唑环是困难的。因此,广泛的理论工作已开展,以确定合成未取代五唑的最佳条件和前驱体[2,24–29]。

图1. cyclo-N5−的合成路线。Route 1显示了中间体ArN5(最常见的取代五唑),但也尝试了其他取代五唑中间体。
2.1. 芳基取代和苄基取代的五唑
最简单的取代ArN5是苯基五唑(phenylpentazole,PhN5)。首次针对这类化合物的实验尝试是由Noelting等[30]在1892年进行的,但该实验只生成了芳基叠氮化物。ArN5的首次成功合成通常归功于Huisgen和Ugi,但实际上应该归功于Clusius和Hürzeler [31]在1954年所做的工作。在1954年之后,Huisgen和Ugi [32,33]能够通过气体体积和动力学研究来检测ArN5是否存在。1983 年,Wallis和Dunitz [34]从物理角度证明了ArN5的结构,他们报道了第一个由X射线测量的4-二甲氨基吡啶五唑(dimethylaminophenylpentazole, DMAP-N5)晶体,Yang等[35]在室温下分解该五唑,以进一步分析该化合物。
PhN5的不稳定性在很大程度上限制了其作为其他五唑前驱体的实验用途。计算和实验研究表明,在苯环上的五唑环的对位和邻位上的供电子基团会使ArN5热稳定,而在相同位置上的吸电子基团则会使ArN5失稳[28,29]。五唑环上的芳基取代基也会使ArN5不稳定。这些结果可以通过芳基与五唑环之间的共振来解释,即稳定性随着共振的增加而增加[28,29]。研究人员发现,取代的ArN5中C–N键的解离能为362~402 kJ∙mol−1,而五唑(N–N键)的解离能为109~117 kJ∙mol−1,说明五唑环在被其取代基裂解之前断裂(图2)[2,29]。Burke和Fazen [25]于2009年对中间体芳基五唑的相同影响进行了探索,Ren等[24]于2019年再次探索了这些影响。这些研究强调了供电子取代基对反式-顺式异构体形成的重要性,反式-顺式异构体是唯一能够成功环化成芳基五唑的五唑中间体。Ren等[24]还提供了可能的五唑中间体的详细反应途径,并描述了取代基对五唑环化作用的影响。

图2. ArN5的分解(单位:Å)。
人们还通过计算探索出在邻位、间位或对位上被单一碱金属锂(Li)、钠(Na)或钾(K)取代的PhN5[36]。研究发现,与Zhang等[29]用–OH、–OCH3和–N(CH3)2取代的PhN5化合物相比,金属取代的PhN5衍生物显著提高了五唑环分解的能垒。同时实验还观察到在邻位金属取代基和氮环之间的金属-氮键。对位和间位金属取代的化合物不存在这种相互作用(图3)[36]。邻位取代的化合物的强分子内相互作用有效地在五唑环上施加了更多的负电荷。负电荷的增加意味着分散相互作用增加,从而导致了五唑环的稳定性更高[36]。
苄基五唑(benzylpentazole, BnN5)及其衍生物也被认为是cyclo-N5−的前驱体[2,37]。Zhang等[37]通过计算研究了吸电子取代基和供电子取代基对五唑环和C–N键稳定性的影响。吸电子基团同时降低了五唑环和C–N键的稳定性。与ArN5前驱体相比,BnN5及其衍生物具有更稳定的N5环和更不稳定的C–N键[28,37]。乍看起来,BnN5似乎是未取代cyclo-N5−前驱体的更好选择。然而,由于必需的苄基重氮合成中间体极度不稳定,BnN5将很难或几乎不可能通过实验产生[38]。
2.2. 杂环取代的五唑
四唑基五唑是第二个被合成的取代五唑[39]。由于它在–50 ℃时分解,因此在-70 ℃进行的核磁共振(nuclear magnetic resonance, NMR)研究是其存在的唯一证明。计算方法表明,四唑对五唑共轭的影响与芳基相似。与中性取代基相比,阴离子型四唑基五唑的分解活化能增加了[39]。
Zhang等[40]也认为六元杂环吡啶五唑(PyN5)及其衍生物是五唑的潜在前驱体。用吡啶基取代PhN5的芳基可降低五唑N–N解离的活化能(8.4~30.8 kJ∙mol–1)和C–N键解离的活化能(11.3~21.0 kJ∙mol–1)[40]。五唑环稳定性的下降比C–N键的下降更为显著。因此,与芳基五唑相比,PyN5并不是cyclo-N5–更好的前驱体[40]。
2.3. 其他的取代五唑
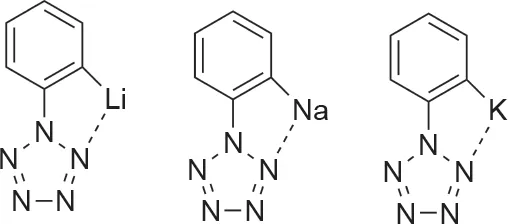
图3. 邻位取代的碱金属Li、Na和K苯基五唑的金属-五唑相互作用[36]。
五唑环的稳定性取决于其芳香性。由于氮-氮三键是最稳定的构象,与双键或单键相比,增加芳香性有助于使环中所有氮原子之间具有更强的双键性,从而提高五唑环的稳定性[41]。分离σ电子和π电子体系的能力也有助于五唑环的稳定[42]。由于这些特性,五唑是一种相对稳定的多氮化合物,可以用作其他多氮体系的结构单元。从n = 5到n = 60的多氮(Nn)体系可以被分为几个不同的组,具体取决于氮原子的数量及其相对构象的稳定性[图4和图5(a)] [8,41,43–46]。通常,含有多个五唑环的结构比相应的开链结构更稳定,如在N18、N20和N30上发现的那样,其中N8叠氮五唑和N10双五唑结构代表了最小能量[图4和图5(a)] [47,48]。必须指出,经过计算,很多人都希望得到的N5+N5–化合物并不存在[图5(a)] [8,47]。

图4. 多氮化合物包含10个氮原子的不同构型。化合物的稳定性从左到右依次降低(化合物来自参考文献[41])。

图5. 其他经过计算探索出的取代五唑化合物。(a)N5基多氮;(b)阳离子取代五唑(R-N5H+);(c)中性取代五唑(R-N5)(化合物来自参考文献[8,43–46])。
根据计算,其他的取代五唑,包括甲基、氰基、氨基和N-氧化物基团五唑都是稳定的高性能化合物[图5(b)~(d)] [43–46,49]。最近还进行了一项对氧平衡为零或更高的五唑衍生物的计算研究(图6)[49,50]。尽管尚无法使用必要的合成方法进行实验合成,但许多这类化合物仍被预测为HEDM的良好候选者。
3.c yclo-N5−的合成方法
到目前为止,只有两种生成cyclo-N5–的方法(图1)。实验上最方便的路线是从芳基取代五唑前驱体上裂解五唑离子[2]。为了使这些前驱体有效,必须考虑以下三个方面:①供电子取代基必须置于苯环相对于五唑环的对位,以便使五唑环有尽可能大的电子密度,从而增加五唑环的稳定性并削弱C–N键的连接;②C–N键必须断裂,而五唑环的N–N键保持完整;③必须使用有效的分析方法检测五唑阴离子。
3.1. 通过电子激发裂解PhN5 C–N 键
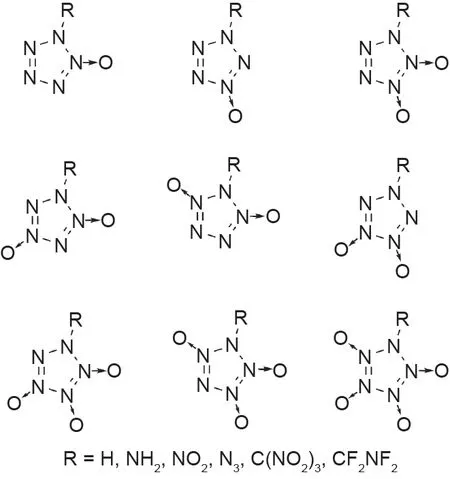
图6. 含N-氧化物的五唑衍生物(化合物来自参考文献[49,50])。
Christe和Östmark进行的质谱研究以及Haas的计算研究揭示了大量有关ArN5系统中C–N裂解的机理。Christe的研究将串联质谱(mass spectrometry, MS/MS)与对羟基和对二甲氨基苯基五唑在高、低碰撞电压下的碰撞诱导解离(collision-induced dissociation, CID)结合使用。在研究对羟基苯基五唑时发现,在低电压下,母体阴离子先分解出N2基团,最后分解出CO。在高电压下,C–N键被裂解成cyclo-N5–,然后分解为叠氮化物阴离子和中性N2(图7)[23]。
Östmark等[51]使用激光解吸电离(laser desorption ionization, LDI)飞行时间(time-of-flight, TOF)质谱技术对DMAP-N5进行了相同的实验,并采用密度泛函理论(density-functional theory, DFT)破译裂解途径。计算表明,通过电子附着产生DMAP-N5自由基,然后分解为cyclo-N5–和DMAP自由基是首选途径(图8) [51]。实验还发现,C–N键断裂的活化能比五唑环的分解活化能高1.6~6.6 kcal∙mol–1[51]。在较高的激光功率下,通过实验观察到,尽管看到了更多的碎片,但在cyclo-N5–分解之前,总是发生C–N键裂解。Östmark等[51]进一步得出结论,可以通过诱导C–N键断裂来激发DMAP-N5,但是这种方法无法生成大量的cyclo-N5–[51]。
Christe和Östmark获得的对比结果通过Belau等[52]的计算研究得到了解释。该研究将闭壳体系的对羟基苯基五唑阴离子与开壳体系的DMAP-N5阴离子自由基等价物进行了比较,发现羟基苯基五唑高能裂解的意外结果可以用反应物和产物的电子态来解释。当C–N键被拉伸时,羟基苯基五唑阴离子负电荷从氧转移到五唑环上,而DMAP-N5阴离子自由基中的负电荷在激发过程中仍保留在五唑环上[52]。该研究还观察到两种物质都存在圆锥形交点,从而允许从激发态形成cyclo-N5–(图 9和图10)。羟基苯基五唑体系仅能在高电压下产生cyclo-N5–,因为产生的激发态能够通过一个圆锥形交点到达相应的基态。但是,DMAP-N5体系的情况并非如此。不同之处在于C–N键裂解和N2挤出的势垒。DMAP-N5阴离子自由基具有类似的势垒,但羟基苯基五唑对C–N键断裂的势垒增加,而对N2挤出的势垒降低[52]。Portius等[53]在使用激光激发和红外光谱电化学研究DMAP-N5的激发态时也得到了同样的结果。

图7. Vij等[23]观察到的对甲氧基苯基五唑的断裂。ESI:电喷雾电离。

图8. 建议的DMAP-N5分解途径,由Östmark等[51]研究。活化能(Ea)与热焓(ΔH)是在298 K下以kcal·mol–1的单位测得的。
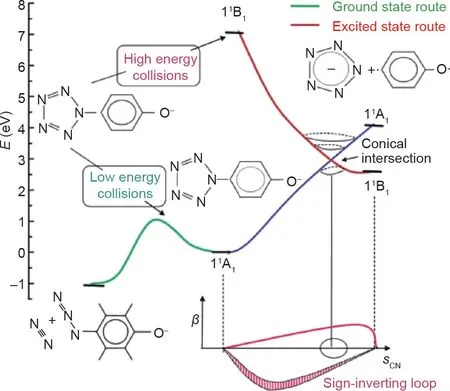
图9. 高、低碰撞电压下对羟基苯基五唑的能级。SCN代表C–N拉伸坐标;β代表向上弯曲(+β)或向下弯曲(–β)。转载自参考文献[52],经美国化学学会许可,©2004。
利用拉曼光谱和紫外-可见(ultraviolet-visible, UVVis)光谱作为分析方法,Haas的小组[54–57]多次尝试用光化学方法来制备cyclo-N5–。他们在不同条件和激发模式下进行了各种实验。发现在所有情况下,如果形成任何cyclo-N5–,则其含量仅为痕量,主要产物是前驱体的分解产物[54–57]。
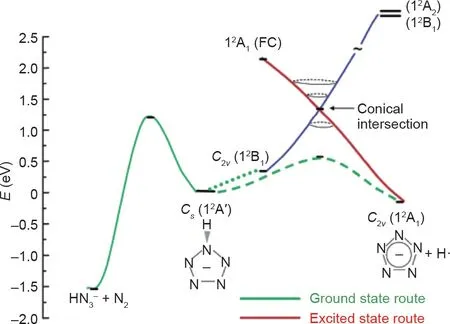
图10. DMAP-N5自由基阴离子等效体系的解离方案。FC代表Franck-Condon激发。转载自参考文献[52],经美国化学学会许可,©2004。
3.2. 氧化还原C–N 键的方法
Butler等[58]尝试使用硝酸铈铵(ceric ammonium nitrate, CAN)、单电子转移(single-electron transfer, SET)试剂和硝酸锌进行ArN5的氧化还原裂解,并获取五唑阴离子。他们报道说,从反应中获得(–10.0 ± 2.0) ppm的独特15N-NMR信号是ZnN5(NO3)。这是发表的有关分离的五唑的首次声明。此主张后来被Schroer等[59]反驳,这促使Butler等用被一个、两个或三个15N原子取代的各种ArN5化合物重复该实验[60]。在这些条件下,Butler 等[60]仅发现了N5–的分解产物。
Bazanov等[12]通过使用碱金属在低温下还原ArN5首次成功地检测到了THF溶液中的五唑阴离子。碱金属能够从母体PhN5中生成PhN5自由基阴离子,然后使其升温至室温以生成cyclo-N5–[12]。通过电喷雾电离(electrospray ionization, ESI)MS检测信号在m/z = 70时的五唑阴离子。MS信号在m/z = 70时,同位素标记一个或两个氮原子产生相应的m/z = 71和72。MS/MS对m/z =70处信号的分析表明,在m/z = 42时出现峰值,对应叠氮化物阴离子(预期的五唑分解产物)。五唑阴离子在低于–40 ℃的温度下储存时是无限稳定的,但在室温下其稳定状态仅能保持几分钟[12]。
Bazanov等[61]后来重复了该实验,以优化反应条件。当在纯溶剂中使用纯金属钠时,五唑阴离子未生成。然而,使用氧化物钝化(固体电解质界面)的钠可产生最高数量的五唑阴离子[61]。不足之处是这种反应方法受到反应时间过长的限制。反应时间最短为两周,最长为3个月。由于反应时间慢且收率仅够进行检测,而不能进行批量分离,因此,Bazanov等[61]给出的结论是钠作为还原剂效用过强了。
Zhang等[13]的实验是通过氧化还原化学生产五唑阴离子最成功的例子。他们在第一个也是最大的批量生产五唑类化合物的实验中,使用双甘氨酸亚铁和间氯过苯甲酸体系以19%的产率从PhN5中裂解了五唑阴离子[13]。最初产生的五唑化合物据说是(N5)6(H3O)3(NH4)4Cl,其中五唑是否发生了质子化,几组人员对此进行了激烈的辩论[62–65]。Huang和Xu [62]认为,在不对称单位图中的氧气椭球似乎表明,所提出的化合物的电子密度小于应分配的值。预期化合物的对称性和所研究的红外光谱(C2v与D5h相比)对称性的不一致性也受到了质疑。DFT计算表明五唑阴离子实际上是由H3O+质子化形成的,并且更可能以HN5的形式存在[62]。Jing等[63]坚持认为,他们的高质量X射线晶体学数据以及NMR和DFT热力学研究能为提出的无质子化五唑提供更准确、更有力的证明。后来Chen等[64]注意到Zhang等没有考虑诸如温度和溶剂之类的外部条件如何严重影响固相和液相中的质子转移。Chen等[64]使用从头算分子动力学(ab initio molecular dynamics, AIMD)模拟来更准确地预测cyclo-N5–在(N5)6(H3O)3(NH4)4Cl晶体和二甲基亚砜(dimethyl sulfoxide, DMSO)溶剂中的变化。他们发现,在固体中cyclo-N5–总是与一定量的质子化的共存物共存,在228 K时最多存在23%的HN5。与之相反,他们发现在DMSO中,溶剂和温度效应缩小了cyclo-N5–与水或氨之间的质子亲和力差异[64]。Huang等[65]关于DFT和AIMD的进一步研究也支持HN5的优先存在。Lu等暗示原始晶体结构的无序性(相关论文尚未发表),导致了众人对它的误解,最终结束了这一争论。他们得出的结论是,生成的化合物实际上是(NH4)7(N5)6Cl,而不是(N5)6(H3O)3(NH4)4Cl [66]。不管辩论结果如何,Zhang等[13]在原始研究中获得的19%的收率迄今为止仍然是五唑盐的最佳收率。产生的盐在室温下也可以保持稳定状态,并在116.8 ℃时分解。
毫无疑问,这项实验的成功,自发布以来的几年间,促使许多其他含五唑的物种合成。不足的是,人们对于C–N键断裂的机理尚未完全了解,这阻碍了更有效的合成方法的发展。在2018年,Yu等[66]通过计算,探索了该实验系统的五唑形成机理,以期可以解决这个问题。尽管他们可以得出结论,即从自由基阴离子转移质子对五唑的形成在动力学上没有什么作用,但他们无法清楚地解释C–N键的裂解。2020年,Shang等[67]提出了裂解五唑阴离子的反应机理。他们得出的结论是,间氯过氧苯甲酸(meta-chloroperoxybenzoic acid, m-CPBA)不负责C–N键断裂的活化,而负责[Fe(Gly)2]的氧化。这种氧化反过来会生成高价的铁(IV)-氧代产物,这是C–N裂解和生成五唑阴离子的关键成分。他们在研究中指出,这种络合物的形成在很大程度上取决于低反应温度,并且当温度过高时,溶液会产生红棕色沉淀[67]。他们得出结论,通过与铁(IV)-氧杂络合物的配体进行实验,可以实现更高的五唑收率[67]。
3.3. N3– + N2 在高温高压下的反应
Oleynik的小组[68–71]执行了另一种目前已知的合成cyclo-N5–的路线,该小组通过在金刚石砧座中加热液态氮和叠氮化物盐,同时激光加热至高温和高压来获取五唑阴离子。此方法可提供足够的能量来克服N3–+ N2环化的势垒,而高压可用于稳定cyclo-N5–。遗憾的是,在释放压力后,没有检测到cyclo-N5–[68–71]。Laniel等[72]进行了类似的实验,他们仅仅能够在惰性气体的环境压力下检测到cyclo-N5–。Laniel等[73]还探讨了由纯锂和过量氮气压缩并加热至最高压力和最高温度分别为73.6 GPa和2500 K的Li-N相图。通过这种方法,他们能够获得Li3N、LiN、LiN2和LiN5,并检查它们各自的压力稳定性域[73]。最近还进行了理论上的高压合成含五唑金属盐的化合物,预测了在环境条件下可以保持稳定状态的铜、镁和钡化合物[74,75]。目前,ArN5前体的裂解仍然是实验室规模制备cyclo-N5–的最佳途径。
4.c yclo-N5−的稳定性和键合
在实验中,已确定五唑阴离子具有C2v对称性,且具有不同的N–N键长。这不同于通过计算方法预测的具有等效N–N键长的D5h对称性[13]。实验发现,氮上的孤对能够与周围的阳离子形成键,从而导致N–N键的长度发生变化。尽管不同的阳离子都可以与五唑环的氮原子结合,所有有记录的键长范围为1.30~1.33 Å,这是处于单键和双键之间的长度。研究还发现,当通过分子内相互作用达到适当稳定时,cyclo-N5–保留了其芳香性 [13,76]。
4.1. 氢键合cyclo-N5–络合物
水合五唑金属盐(钠、锰、铁、钴、镁和钡)的稳定性在实验上表现出对水的严重依赖性[13,77]。最近的一项计算研究也发现,水在稳定水合五唑金属盐中起着重要作用[78]。这是由于水既可以充当氢键供体又可以充当受体[12,78]。在所有情况下,水分子要么通过与金属的配位(配位水),要么通过与五唑阴离子形成氢键(配位水),来稳定络合物(图11)[78]。
与水分子上孤对的相互作用导致金属4s和3d轨道的能量增加,并随后形成导数(或配位)键[78]。反过来,通过降低金属的吸电子效应和增加N2挤出的活化能而降低金属与五唑阴离子的相互作用。实验还观察到水在金属-五唑络合物上会引起位阻,从而限制五唑阴离子的配位方向[13]。
与配位水相反,结构水与金属-五唑络合物中的两个单独的cyclo-N5取代基的氮原子形成氢键(图11)。从金属-五唑络合物中除去结构水分子后,N2挤出的活化能显著降低。因此,失去水分会导致化合物不稳定,这是分解的第一步[78]。必须注意的是,实验发现配位水分子对水合五唑金属盐稳定性的影响比配位水更重要[78]。在该研究中,配位水导致分解的动力学势垒平均增加7.6 kcal·mol–1,而结构水导致分解的动力学势垒平均增加7.3 kcal·mol–1[78]。
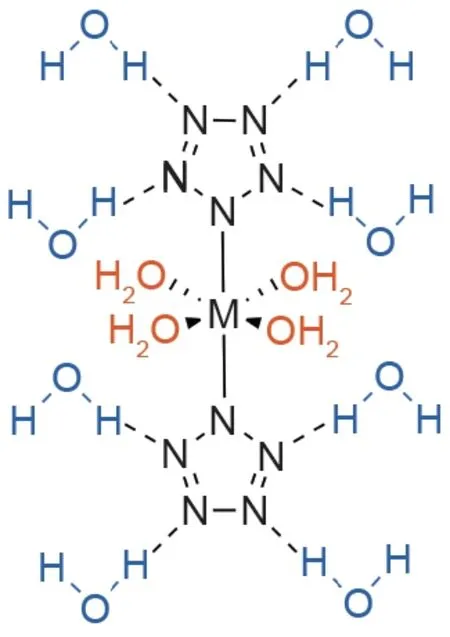
图11. 水合五唑金属盐中配位水(橙色)和结构水(蓝色)的键合模式。
由Xu等[19]合成的不含金属的有机盐是五唑阴离子替代水与氨基形成氢键,生成的无水盐。应该注意的是,产生的所有盐都与每个五唑形成氢键。研究还发现,这些氢键比分子中发现的总范德华力更强,从而使它们成为化合物具有稳定性的重要因素[19]。Xu等[79]还生成了无金属的水合五唑盐(C4H9N10+)(N5–)·3H2O和(C5H9N10+)2(Cl–)(N5–)·3.5H2O,它们都具有N–H和O–H氢键。当比较两种化合物的热稳定性和Hirshfeld表面分析时,N–H氢键比O–H氢键更有利于cyclo-N5–环的稳定 [79]。
4.2. cyclo-N5–的酸度和稳定性
Zhang等[80]的量子计算发现,五唑阴离子同时具有σ和π芳香性。π芳香性是由环状氮原子的杂化产生的,而孤对能够在五元体系中扩散,从而产生σ芳香性。应该注意的是,这种“双重芳香性”与金属芳香族体系中观察到的不同。在cyclo-N5–中,这些芳族体系是分开的,而在金属芳族体系中,它们是结合的[80]。这些竞争性的芳族体系稳定了cyclo-N5–。
研究还观察到,五唑阴离子的双芳族体系受溶液中酸摩尔比的影响。五唑阴离子与一个和两个水合氢离子结合会降低系统的芳香性,使其稳定性降低。但是,如果可以在溶液中保持3~4个摩尔当量的水合氢离子,则五唑阴离子能够克服酸碱中和规则,并形成氢键络合物以恢复其双重芳香性[80]。通过这种方式,酸性环境可以使cyclo-N5–稳定。此特性很可能是Zhang等[13]开发的双甘氨酸亚铁和m-CPBA裂解以及稳定系统之所以如此成功的原因。因为之前的其他合成方法都不是酸性反应溶液。
4.3. 金属cyclo-N5–化合物中的键合
多年来,人们已经对各种金属阳离子与cyclo-N5–形成络合物的能力进行了研究[11,81–91]。已计算出的金属-五唑络合物(五唑盐)具有三种可能的配位模式:单齿状(η1-N5)、双齿状(η2-N5)和类茂金属(η5-N5)。单配体和二齿配体涉及五唑阴离子的σ键,而类茂金属的配位涉及π键(图12)[11]。在碱金属(Na和K)和碱土金属(Mg、Ca、Sr和Ba)的五唑盐中,通过计算观察到η2-N5配位,而且大多数络合物是平面的[84,85]。锂是唯一异常值,因为它更倾向于η5-N5的配位形式 [11]。人们对几种过渡金属和稀土金属的五唑盐也进行过计算研究,对于含单和双五唑的化合物,同时观察到η2-N5和η5-N5的配位。此外,研究还发现η1-N5对于多五唑结构更有利[86–89]。金属-五唑络合物的键合模式的实验结果与所计算值不同。在与已通过实验生产的金属-五唑络合物进行比较时,仅观察到五唑阴离子以单齿方式进行配位,其中环中的任何一个氮原子至所有5个氮原子均可形成单齿键,最多可形成5个分离的金属阳离子[92]。实验中还没有观察到双齿和类茂金属的结构[13–18,81,93]。
5. 含能五唑盐化合物
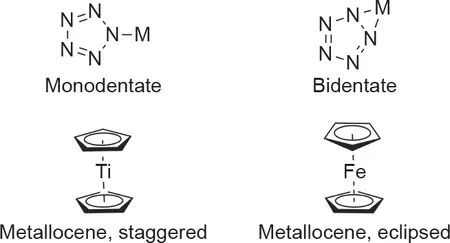
图12. 五唑盐的配位模式。
计算研究已经预测了含有五唑盐的化合物的许多稳定形式,并且还试图预测其作为高能材料的性能[8,45–50,94]。这类研究是无价的,因为它们通常有助于指导实验人员进行综合设计,并且在某些情况下,还可以进一步佐证实验结果(如Christe等[94]的最新工作)。尽管计算研究远远超过了实验研究,但在过去几年已通过实验获得了几种类型的高能五唑络合物。因为密度、生成热和分解温度都已确定,所以这些化合物中的大多数都作为高能材料被检测。利用这些性质,人们已经通过计算预测了几种五唑盐的爆炸性能(即爆轰压力和速度)。基于五唑的配位聚合物(coordination polymer, CP)也已经通过实验获得[81]。在大多数情况下,由于金属-无机骨架(metal-inorganic framework, MIF)的密度低,因此相比于作为高能粒子,它们更适用于研究五唑的键合模式。这些材料中的大部分因其爆轰速度或敏感度而没有得到进一步表征。由于对这些化合物的爆轰参数最感兴趣,因此我们仅讨论被充分表征为高能材料的化合物,包括其爆轰速度、压力和敏感性(图13)。表1总结了所有讨论的化合物的性质[2,16,19–22,81,96]。
5.1. 热稳定性
在报道的这些化合物中,三维(3D)MIF化合物17具有最高的分解温度(129 ℃)。其结构表明,五唑环中的平均N–N键长度为1.323 Å,长度介于单键和双键之间[16]。化合物中的每个五唑氮也与钠离子键合,5个配位的钠离子几乎与五唑环共面[16]。由钠、五唑和氧离子配位产生的3D结构包括环应变、螯合、桥联作用和静电力,这归因于相对于含五唑的化合物的高热稳定性[16]。
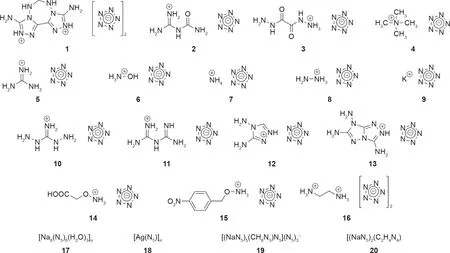
图13. 表1中所述的含五唑的高能化合物。1~16:高能盐;17、18:三维(3D)MIF;19、20:3D CP。化合物17~20的结构可在表1列出的相应参考文献中找到。

表1 含能五唑盐化合物的能量性质
含能五唑非金属盐取决于抗衡阳离子和氢键的热稳定性[20]。对于这类化合物,在进一步分解发生之前,五唑环的分解几乎总是伴随着HN3气体的形成。这种进一步的分解取决于抗衡阳离子[20]。化合物11具有所有含能五唑非金属盐的最高分解温度(124.8 ℃)[21]。五唑环中的每个氮原子都与抗衡阳离子形成氢键,每个抗衡阳离子与两个相邻的五唑环之一接近共面[21]。该化合物相对较高的热稳定性归因于其广泛的氢键作用[21]。具有相对最低的热稳定性的化合物是化合物4,其分解温度为80.8 ℃ [20]。该化合物缺乏氢键合能力和配位键[20]。通常,大多数五唑盐在110 ℃以下会分解。
5.2. 机械感度
通常,机械感度(即对摩擦和冲击的敏感度)取决于化合物内的分子间相互作用[3]。机械感度随着氢键的增加和固态下的逐层堆积而增加,因为这些相互作用能够吸收机械能,并将其转化为动能,以层滑动的形式出现[21]。化合物5、12和13对冲击和摩擦不敏感[冲击感度(impact sensitivity, IS)> 40 J,摩擦感度(friction sensitivity, FS)> 120 N] [21]。这取决于它们的逐层堆叠模式,其中各层之间存在π-π相互作用[21]。还应注意的是,在化合物5、12和13的各层之间观察到的氢键和π-π相互作用比其他化合物更强[21]。与其相反的是,化合物9和18缺乏这些功能,因此对机械刺激最敏感。一般而言,含五唑盐的化合物比传统炸药(如TNT、RDX和HMX)的机械稳定性更强(表1)。
5.3. 爆轰性能
爆轰性能在很大程度上取决于分子的密度和生成的热量[20]。本文所提供的20种化合物均具有相对较低的密度。仅有6种具有超过TNT的密度(1.65 g·cm–3),但是与传统炸药(–59~80 kJ·mol–1)相比,因为所有化合物的氮含量有所增加,所以它们的生成热都很高(95.2~1362.4 kJ·mol–1)(表1)。据此,可以得出结论,含五唑的化合物的性能将在很大程度上反映生成热。此外,在不断寻找新的高能五唑时,应将重点放在具有高密度潜力的化合物上。高密度五唑与先天形成的五唑阴离子的高热量相结合,将使材料具有非常高的性能。化合物3、6、8和11的爆轰速度最高,且均超过HMX。化合物11的有趣之处在于,其生成热是双胍高生成热(1618 kJ·mol–1)的结果,这表明通过引入高生成热的阳离子可以增加含五唑的化合物的生成热[21]。与富含氮的五唑盐化合物相比,五唑金属盐化合物通常具有较高的密度,但性能较低。例如,化合物18具有3.105 g·cm–3的极高密度,但其爆轰速度仅为7782 m·s–1,因为大部分密度来自缺乏能量含量的银阳离子。在氮含量较低的五唑非金属化合物(如化合物4)中,可以看到相同类型的低性能,其爆轰速度为5880 m·s–1。在上面显示的20种化合物中,有13种的爆轰速度高于TNT。
6. 结论
最近,实验室有效地从ArN5中规模制备五唑阴离子,使得大众对五唑阴离子及其衍生物的兴趣大大增加。基于数十年对计算预测的兴趣,这一进展最终使高能五唑领域的研究得以确立并蓬勃发展,目前已知的五唑阴离子的许多高能衍生物已被充分表征。五唑的生成热极高,因此预示其未来将会被用于高能材料。然而,在运用五唑之前,人们需要具有更高热稳定性的衍生物。实验工作之前进行的计算工作在指导合成工作和基于五唑的新型高能材料的开发方面具有不可估量的价值,这佐证了实验者和理论家之间可能产生的生产协同作用。
Acknowledgements
We thank the grant from Army Research Office (ARO)(W911NF-18-1-0463), the Office of Naval Research (ONR)and Purdue University for the support of our lab.
Compliance with ethics guidelines
Dominique R. Wozniak and Davin G. Piercey declare that they have no conflict of interest or financial conflicts to disclose.
猜你喜欢
高中数理化(2022年14期)2022-08-15
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
原子与分子物理学报(2019年5期)2019-04-28
当代陕西(2019年6期)2019-04-17
中学化学(2015年12期)2016-01-19
现代农业(2015年3期)2015-02-28
外语学刊(2014年3期)2014-12-03
应用化工(2014年1期)2014-08-16
原子与分子物理学报(2014年3期)2014-02-28
郑州大学学报(理学版)(2013年2期)2013-03-11
