
微生物细胞工厂的设计构建:从诱变育种到全基因组定制化创制
2021-01-21 01:55袁姚梦邢新会张翀
合成生物学 2020年6期
袁姚梦,邢新会,2,张翀
(1 工业生物催化教育部重点实验室,清华大学化工系生物化工研究所,清华大学合成与系统生物学研究中心,北京100084; 2 清华大学深圳国际研究生院生物医药与健康工程研究院,广东 深圳 518055)
以石油等不可再生化石资源为原料的传统制造业面临着“高能耗、高污染、高排放”等严峻挑战,亟需通过生产原料、加工过程和产品创制的绿色变革,实现未来的可持续发展。绿色生物制造以可再生资源为原料,利用生物催化和转化过程,实现高附加值产品的绿色制造,由于其能够实现工业制造模式从“末端治理”到“源头控制”、从“低端产品”到“高值化转化”的转变,成为推动生物经济发展的重要路径之一。
微生物细胞工厂(microbial cell factories,MCFs)是绿色生物制造的核心环节。利用微生物强大且多样的生化反应网络,通过对代谢路径的重塑和工程化,可以将微生物细胞改造为能够以低价值可再生资源为原料生产各类产品的MCFs。从青霉素[1]、谷氨酸[2]、乙醇、1,3-丙二醇[3]、法尼烯[4]到阿片类药物[5]等生物合成 MCFs 的成功开发,展示了MCFs 合成高附加值化学品的巨大潜力。迄今为止,MCFs 已能够生产抗生素、氨基酸、重组蛋白、生物能源[6]、生物塑料乃至“人造肉”,被广泛应用于制药、食品、能源和农业等领域[7-12]。
MCFs 的构建策略经历了不同的历史阶段。在20 世纪90 年代之前,主要通过天然微生物的筛选和非理性诱变育种技术获得目标产物高产菌株。这种将随机突变和定向筛选相结合的策略在工业菌株的开发上已经有诸多经典成功案例[13-15]。然而,由于突变过程的随机性,这种策略往往花费时间长、工作量大,是一种典型的“以时间(人力)换水平”的策略。尽管如此,由于其具有应用历史悠久、操作简单、适用范围广、属于非转基因操作等优势,非理性诱变育种策略至今仍然是微生物育种研究和产业应用的常用平台技术。
20 世纪90 年代以来,随着分子生物学、基因工程技术的逐步引入,代谢工程学科正式创立[16-17]。代谢工程利用重组DNA 技术对生物体中已知的代谢途径进行有目的的设计,以更好地理解和利用细胞途径,并对细胞内的基因网络和调节过程进行调控和优化,构建具有特定功能的MCFs,例如,提高现有产品的产率,生产新的产品和扩大可用的底物范围[16-18]。按照代谢工程的理论框架,所构建的MCFs 需要满足[19]:第一,通过对代谢网络物质流和能量流的设计,实现底物消耗和目标产物生产的最优化;第二,改造后的MCFs 能够适应工业生产环境,满足工业生产需求。
随着分子生物学和反向遗传工程手段的发展,人们对于微生物代谢网络及其调控机制的认识得到了巨大的进步,然而,由于微生物系统的复杂性,仍然不能完全理性地设计满足工业需求的MCFs。目前,利用代谢工程策略开发MCFs 的流程主要依靠“设计-构建-测试-学习”(design-buildtest-learning, DBTL)循环[20],通常从MCFs概念设计(proof of concept)到满足实际应用,需要50~300人年和数亿美元的投入[21],耗时耗力、投入高。在代谢工程MCFs 创制的DBTL 循环中,“理性设计”始终是人们的理想目标,也由此应运而生诸多代谢网络模型和设计策略。然而,经典代谢工程指导的设计方法大多都基于已知的生物学知识,由于微生物代谢网络中存在诸多可能对目标表型产生影响的未知因素,或称为“生命暗物质”,这一手段仍然存在诸多局限[22-25]。生物信息学和各种组学技术的快速发展,使得研究者逐渐有能力从系统代谢工程的层次思考MCFs的快速创制,例如,基于全基因组改组技术[26]、新型诱变技术[27]、单敲除库筛选技术[25]、基因组文库共存/共表达(coexisting/coexpressing genomic libraries, CoGeLs)[28]等技术,结合组学和生物信息学手段获取生物学知识,以进一步指导MCFs 的设计。这一理性/半理性结合的策略为突破知识局限,获取更为高效的MCFs 提供了重要的手段,然而,系统代谢工程手段获取知识的效率不高,MCFs 改造过程仍然需要耗费大量的时间和精力。
近年来,合成生物学的进步使得MCFs构建和测试的能力得到显著提升,为提高构建效率以满足市场快速变化和多样的需求提供了重要的机遇。另一方面,二代测序(next generation sequencing,NGS)和基因组编辑的技术飞跃,使得从全基因组层次设计和构建MCFs成为可能。利用高通量研究技术,目前已经可以从全基因组层次并行研究微生物特定表型与基因型的关系,从而获得大规模的基因型-表型关联(genotype phenotype associations,GPA)数据集[29]。如果能够利用这些大规模GPA 数据集,基于数据科学手段从全基因组范围深度挖掘传统分子生物学手段无法发现的未知关联基因及其位点,将有可能从数据(学习)的角度绕开理性设计的知识瓶颈,为提高MCFs设计和创建效率提供全新的研究范式。此外,由于上述数据驱动的全基因组规模定制工程策略基因型搜索范围更宽(全基因组),不依赖于现有知识(数据驱动),将有可能探索之前理性/半理性所无法达到的表型“高地”,获得生产效率更为高效、生产性能更加优越的下一代定制化MCFs。
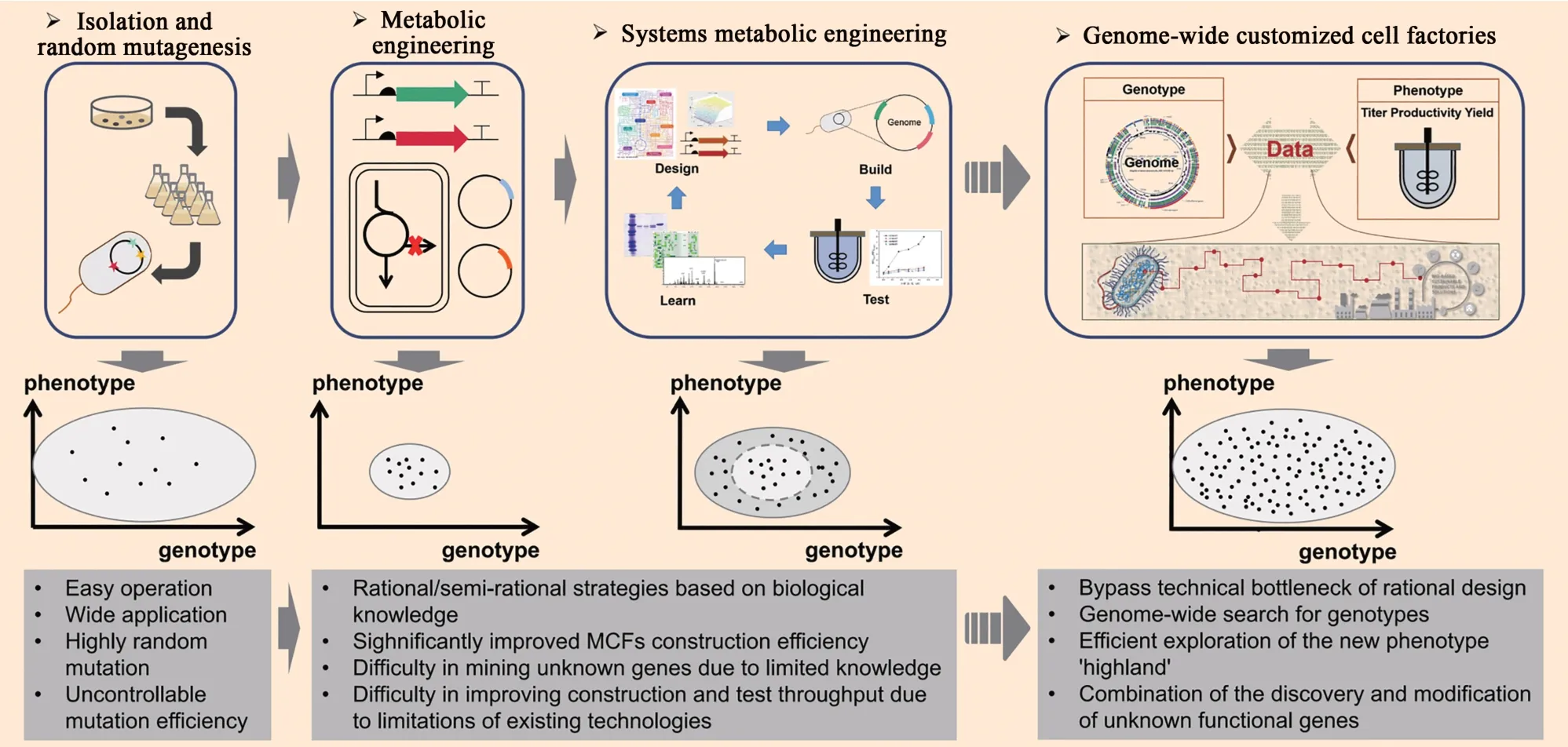
本文将结合实际案例对MCFs的设计及构建策略进行综述,首先回顾传统诱变育种和代谢工程指导的理性/半理性设计策略,接着探讨如何突破代谢工程经典框架限制,利用全基因组规模定制工程实现全基因组水平定制化MCFs的快速构建,最后将对这一新的MCFs 构建范式的未来进行展望(图1)。
1 诱变育种技术在MCFs构建中的应用
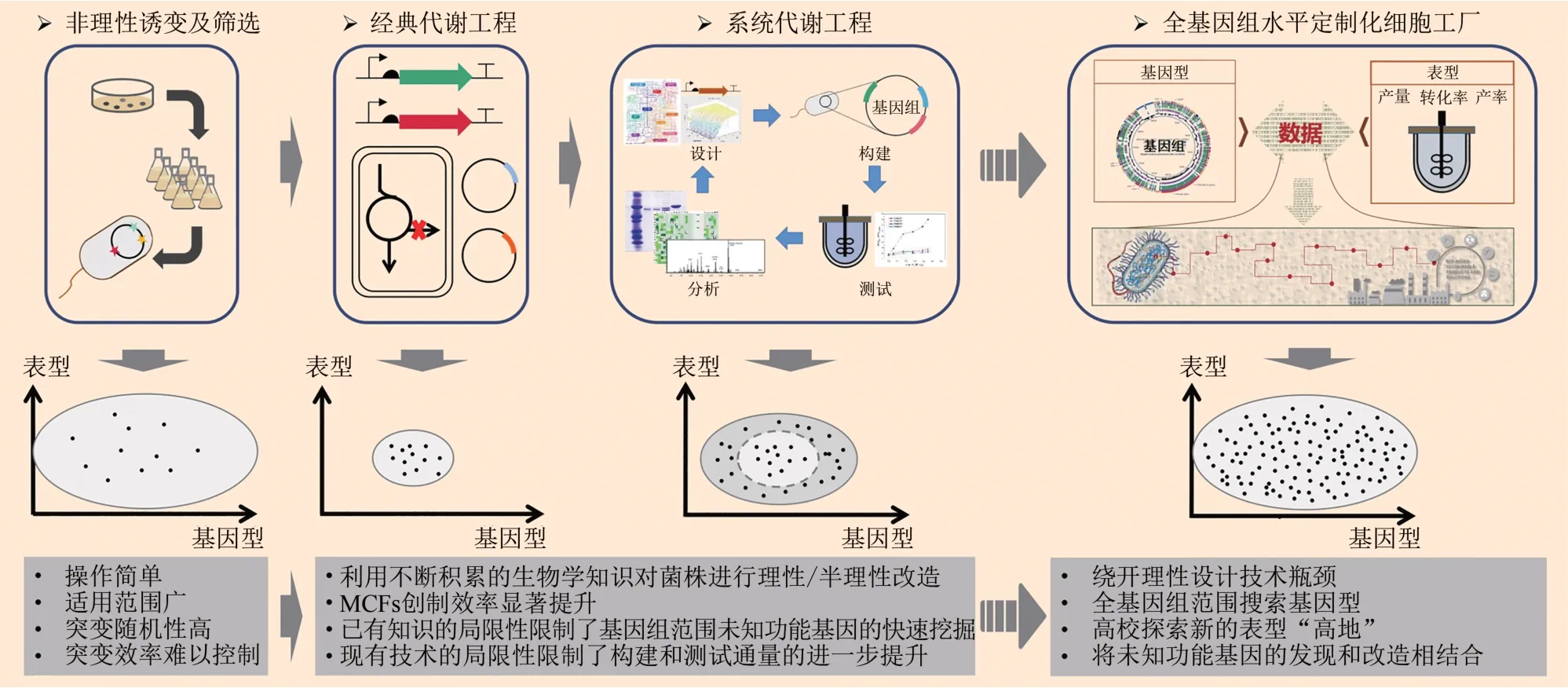
图1 微生物细胞工厂(MCFs)设计和构建发展历程及未来展望Fig.1 Development and future prospects of MCFs design and construction
诱变育种(mutation breeding)是在人为条件下,利用物理、化学、生物因素,诱发生物体产生突变,从中选择、培育植物和微生物新品种的方法,长期以来在科学研究和生物产业中得到广泛应用。诱变育种手段通常包括物理诱变、化学诱变和生物诱变[30](表1)。物理诱变主要采用电离辐射和非电离辐射等物理因素诱发变异;化学诱变主要利用烷化剂、碱基类似物、移码诱变剂、脱氨剂和羟化剂等化学物质诱发变异;生物诱变主要包括噬菌体、质粒、DNA 转座子诱变和原生质体融合、DNA 改组、基因组重排等能够显著提高基因重组频率的诱变技术。诱变产生的突变随机性大、且在全基因组范围分布稀疏,存在大量的无义突变,因此,采用诱变育种技术获得理想性状的微生物往往花费时间长、工作量大,是一种典型的“以时间(人力)换水平”的策略。尽管如此,由于诱变育种技术采用非理性的手段,理论上不需要任何先验的微生物代谢网络结构及其调控的知识,对于没有基因操作手段的微生物,或者具有非转基因(non-GMO)需求的应用领域(如食品),在20世纪90年代之前一直被作为MCFs开发的最重要的手段,目前也仍然是微生物技术科研和发酵产业最为常用的育种手段之一。一个典型的例子是青霉素菌株的选育,1943年,研究者从霉甜瓜中分离得到一株产黄青霉NRRL-1951,其青霉素产量为60 mg/L,在长达50年的人工选育后,产黄青霉的青霉素产量已经达到70 g/L[31]。
近年来,多种新型诱变技术的发展也为诱变育种注入了新的活力。例如离子注入诱变、等离子体诱变等[30](表1),这些新的诱变技术具有突变率高、变异范围广、变异稳定等优点。离子注入诱变要求严格的真空环境和安全防护措施[30],其推广使用受到一定程度的限制。近年来新发展起来的常压室温等离子体诱变技术(ARTP)作为新型物理诱变方法,具有突变率高、突变速度快、突变库容大、安全性高、可控性好、操作简单等优点,在微生物诱变育种中得到广泛的应用[27]。
2 代谢工程指导的理性、半理性设计策略构建MCFs
20 世纪90 年代,随着分子生物学的发展,微生物代谢网络的物质流、能量流以及复杂的调控机制得到了更为深入的研究,基于生物学知识的理性设计作为代谢工程指导的经典设计策略逐渐被应用于 MCFs 构建领域。James E.Bailey[16]最早提出对代谢网络进行理性设计,并利用基因工程的手段对其进行改造以实现代谢流量分布的优化和目标产物产量的提升的设想。Stephanopoulos[17-18]进一步指出,微生物的代谢网络中具有诸多“刚性”节点,能够通过复杂的调控机制抵抗遗传扰动带来的通量变化。从代谢网络分析的角度看,代谢网络的理性设计面临三个挑战[18]:
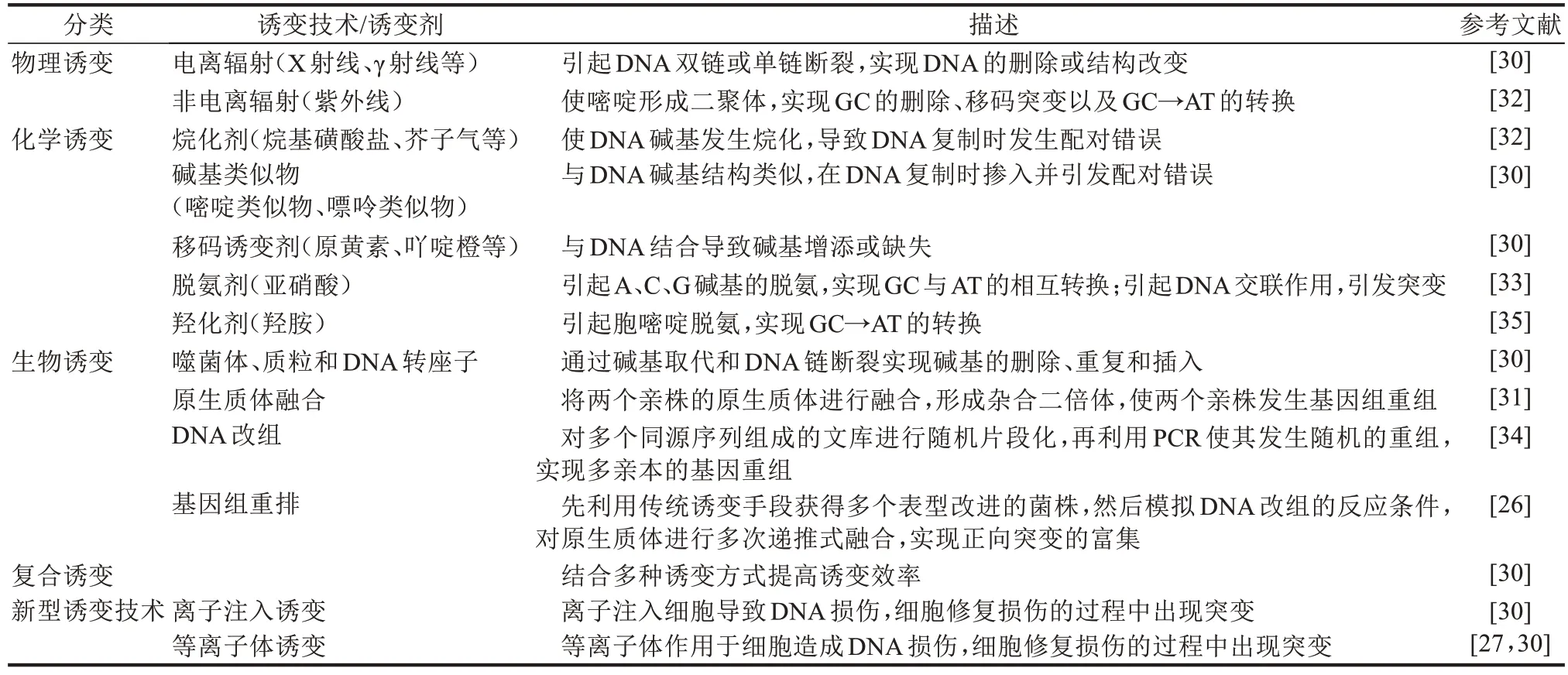
表1 常见物理、化学、生物诱变技术汇总Tab.1 Summary of mutagenesis technologies
(1)识别代谢网络中流量分配显著影响产品合成的关键节点。多数工作仅调控位于产物合成途径末端的反应,取得的成就十分有限。事实上,来源于中心代谢的合成前体、辅因子和能量供应对产物合成也具有重要的作用,识别其中的关键节点是显著提高优化效率的基础。
(2)确定关键节点处最合适的基因改造类型。包括过表达目标产物合成途径、抑制副产物生成酶活性、解除刚性关键节点的调控机制等。代谢网络的流量分布需要严格而小心的调控,确定合适的流量调控类型和程度,是优化MCFs的必要步骤。
(3)优化中心代谢流量分布之后。需要识别产品合成途径中所有的代谢控制位点并对其进行精准调控,同时对潜在竞争途径中的酶活性加以平衡,以获得产量提升、性能稳定的MCFs。
在经典代谢工程领域,多种代谢网络分析和设计方法的不断提出和完善为代谢网络的理性设计提供了理论和技术支撑(表2)。这些分析方法主要包括:动力学分析法,可用于细胞内酶促反应动力学模型的建立和分析;代谢网络分析方法,可用于确定某一目标函数下代谢通量的唯一解或进行途径分析,包括代谢通量分析、通量平衡分析和代谢途径分析三类分析方法;整合了热力学的代谢网络分析,即以热力学第二定律对所有代谢反应加以约束,例如网络嵌入的热力学分析(NET)。随着计算技术的进步和更多代谢模型的建立,多种应变优化算法及软件得到逐步开发和应用,例如基于约束的重构与分析(COBRA)。
在此基础上,研究者逐步阐明了大肠杆菌、谷氨酸棒杆菌、酿酒酵母等微生物的核心代谢网络,并提出了一系列可用于通量平衡分析的化学计量学模型。早期代谢工程领域的代谢途径设计几乎都基于核心代谢网络模型,随着系统生物学的发展,基因组规模代谢重构(genome-scale metabolic reconstruction, GSMR)的概念最先在大肠杆菌中被提出[50],随后,基因组规模代谢模型(genome-scale metabolic model,GSMM)的概念进一步推动了大量基因组规模实验数据与数学建模的结合,增进了人们对于基因组规模代谢的理解[51-52],MCFs 的代谢模型构建规模也逐渐从核心代谢规模上升到基因组规模。Nielsen 和Palsson课题组首次提出了酿酒酵母的代谢模型,这也是真核生物第一个基因组规模代谢模型[53]。随着NGS 技术、组学分析技术的快速发展,越来越多的微生物代谢模型被不断提出和完善(表3)。
代谢网络全基因模型及其计算机优化模拟算法的开发极大地提高了人们对代谢网络的设计和分析能力,这种从系统层次对微生物代谢网络进行理性设计的系统代谢工程方法论逐渐成为MCFs改造领域的研究热点。2011 年,Becker 等[73]通过基因组规模的计算模拟,仅通过12 处基因改造就将谷氨酸棒杆菌内的代谢流重新定向为合成L-赖氨酸的最优途径,该基因工程改造菌LYS-12 生产的L-赖氨酸滴度达到120 g/L,转化率为0.55 g氨基酸/g葡萄糖,产率达到4.0 g/(L·h),以上指标均达到了过去50多年通过诱变育种获得的最高水平,是第一个可与已有工业生产菌株竞争的基因工程设计的L-赖氨酸高产菌。然而由于模型预测准确性还亟待提高,基于该方法论的菌株开发策略仍然依赖于“设计-构建-测试-学习”的循环迭代(试错)流程[20](图2):首先利用系统生物学工具建立微生物的代谢模型,确定MCFs改进目标;接着利用基因工程手段进行菌株构建;对菌株进行表征,并结合高通量分析或组学分析等手段对目标参数进行评估;根据分析结果,对模型进行改进;重复迭代,直至获得满足需求性状的目的MCFs。需要注意的是由于生物系统的多层次网络结构及复杂性,基于系统层次的代谢网络模型进行MCFs开发必须依赖已有的生物学知识。Sang Yup Lee 等[74]主张将进化代谢工程方法论也纳入系统代谢工程方法论体系,通过将理性设计和非理性的随机突变或适应性进化相结合,利用比较组学手段可以获取与微生物代谢网络结构及其调控相关的未知信息,从而进一步提高MCFs设计效率和性能。

表2 用于代谢网络设计的分析方法Tab.2 Analytical methods for metabolic network design
理性/非理性设计指导的MCF 代谢工程改造策略在实际应用中已经取得巨大成功,表4汇总了其中一些代表性案例。然而,由于生物系统的复杂性和“生命暗物质”的普遍存在,目前代谢工程主流采用的“设计-构建-测试-学习”迭代试错流程,通常从MCFs 概念设计(proof of concept)到满足实际应用需求,需要50~300人年和数亿美元的投入。近年来,市场快速变化且多种多样的需求给MCFs 的开发效率提出了更高的要求。另外,基于理性/非理性的设计策略主要基于已知生物学知识,难以达到某些未知的表型“高地”,从而制约MCFs性能的进一步提升。因此,亟需开发更为高效的工程化策略以满足未来快速定制化创制MCFs的需求。
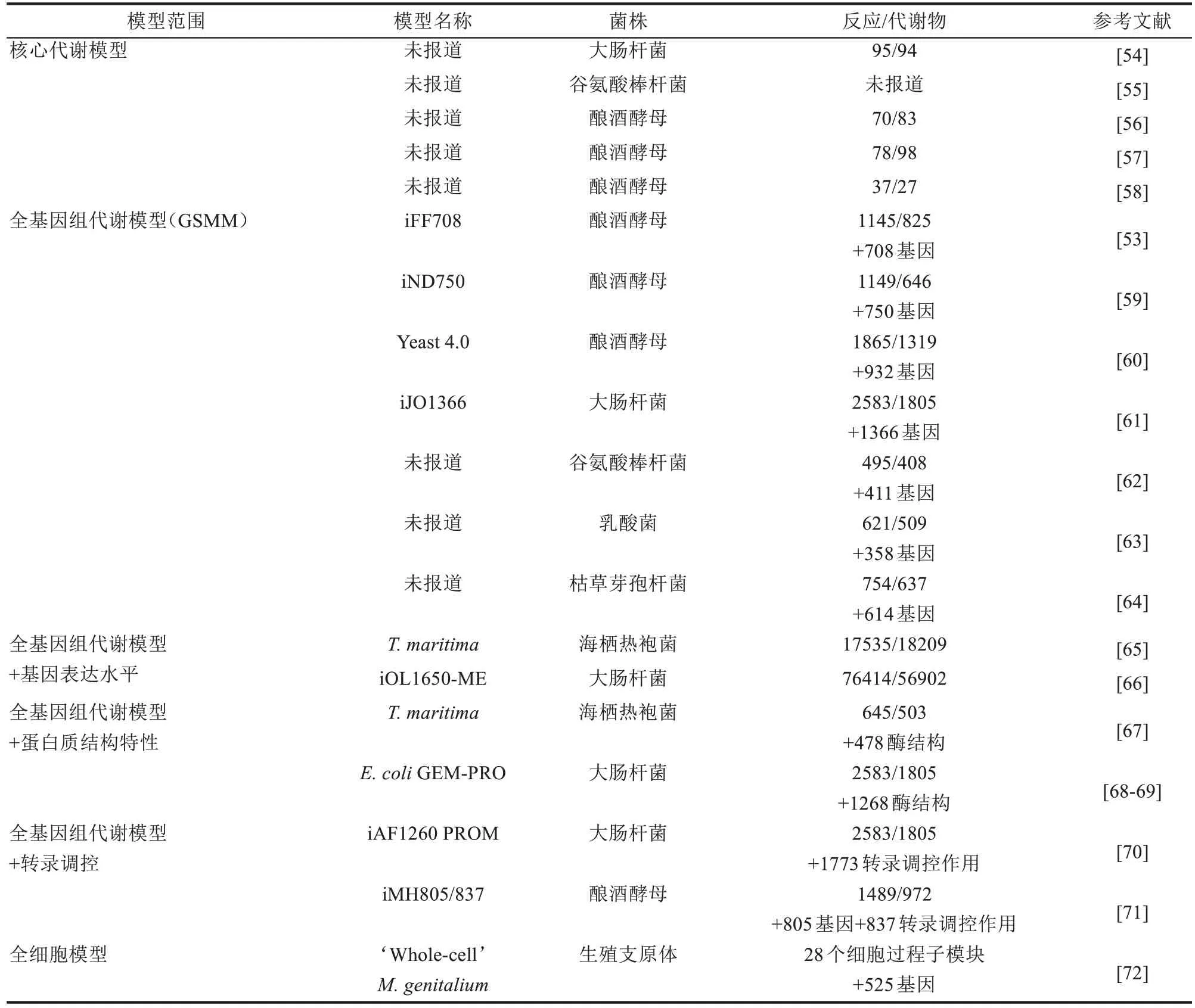
表3 常见的微生物代谢模型汇总Tab.3 Summary of microbial metabolism models
3 全基因组规模定制工程策略构建MCFs
微生物代谢及其控制是一个非线性复杂网络系统,细胞依靠其精巧的基因线路以及严格的调控机制来维持各项代谢活动的稳定。按照“设计-构建-测试-学习”的框架,基于已有的知识,利用基因、启动子、核糖体结合位点等分子元件能够自下而上地在微生物细胞中进行基因线路的设计和构建,从而可望开发出具有特定功能的MCFs。然而,由于已有生物学知识的局限性,生物系统存在极高的不可预测性,导致设计的基因线路在实际体系中难以达到理想的目标。
事实上,随着生物信息学和各种组学技术的快速发展,基因组范围内大量的未知功能基因位点逐渐被人们所认知,包括潜在的别构调节区域[22]、复杂的转录调节网络[23]、功能未知的基因[24-25]等。这些未知因素及其背后的生物学机制限制了现有基于知识的理性/半理性设计的MCFs工程化策略的进一步发展。为了在MCFs基因组范围内挖掘对目标表型有重要影响的基因位点,系统代谢工程领域常利用非理性诱变及筛选手段获得缺陷型菌株,再人为选定目的基因并对其测序,从而找到关键位点。同时,随着分子生物学技术的发展,亦可通过单敲除库筛选[25]、基因组文库共存/共表达[28]、全基因组ORF表达水平调节[86]等技术的发展使得人们能够挖掘基因组上功能未知的基因位点。例如,目前已经在大肠杆菌[87]、枯草芽孢杆菌[88]等模式微生物中对所有基因进行逐一敲除,形成包含数千个不同突变体的单基因敲除菌株阵列(single-gene knockout mutants array),为全基因组范围的功能基因组学研究提供了黄金标准。然而,这些技术往往耗时耗力,成本高昂,且仅能研究全基因范围内少数基因与目标表型的关联(genotype phenotype associations,GPA),亟需通过新技术的引入大幅提升未知功能基因挖掘的效率。
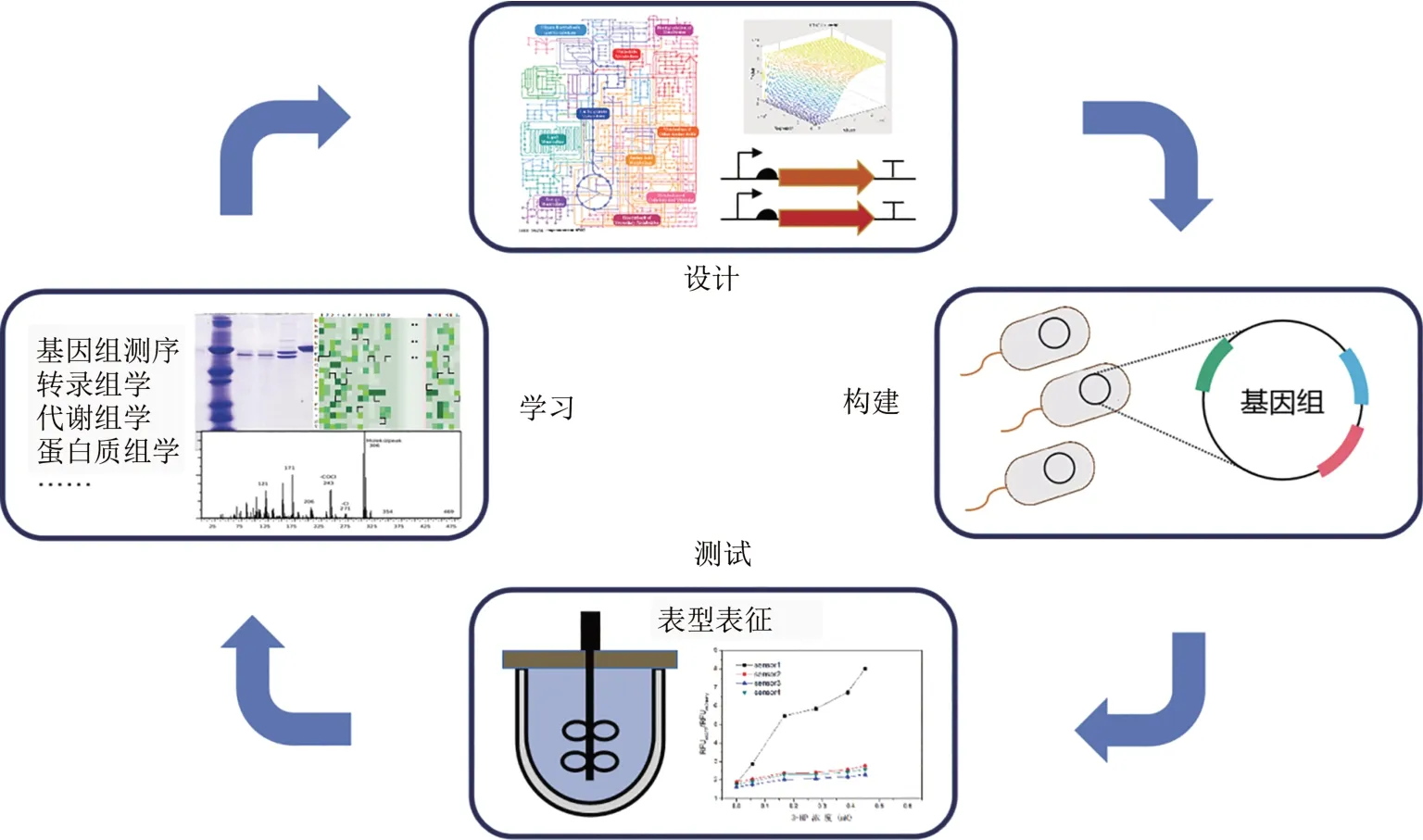
图2 菌株理性工程化的试错流程Fig.2 Ⅰterative trial-and-error cycle of rational engineering of strains
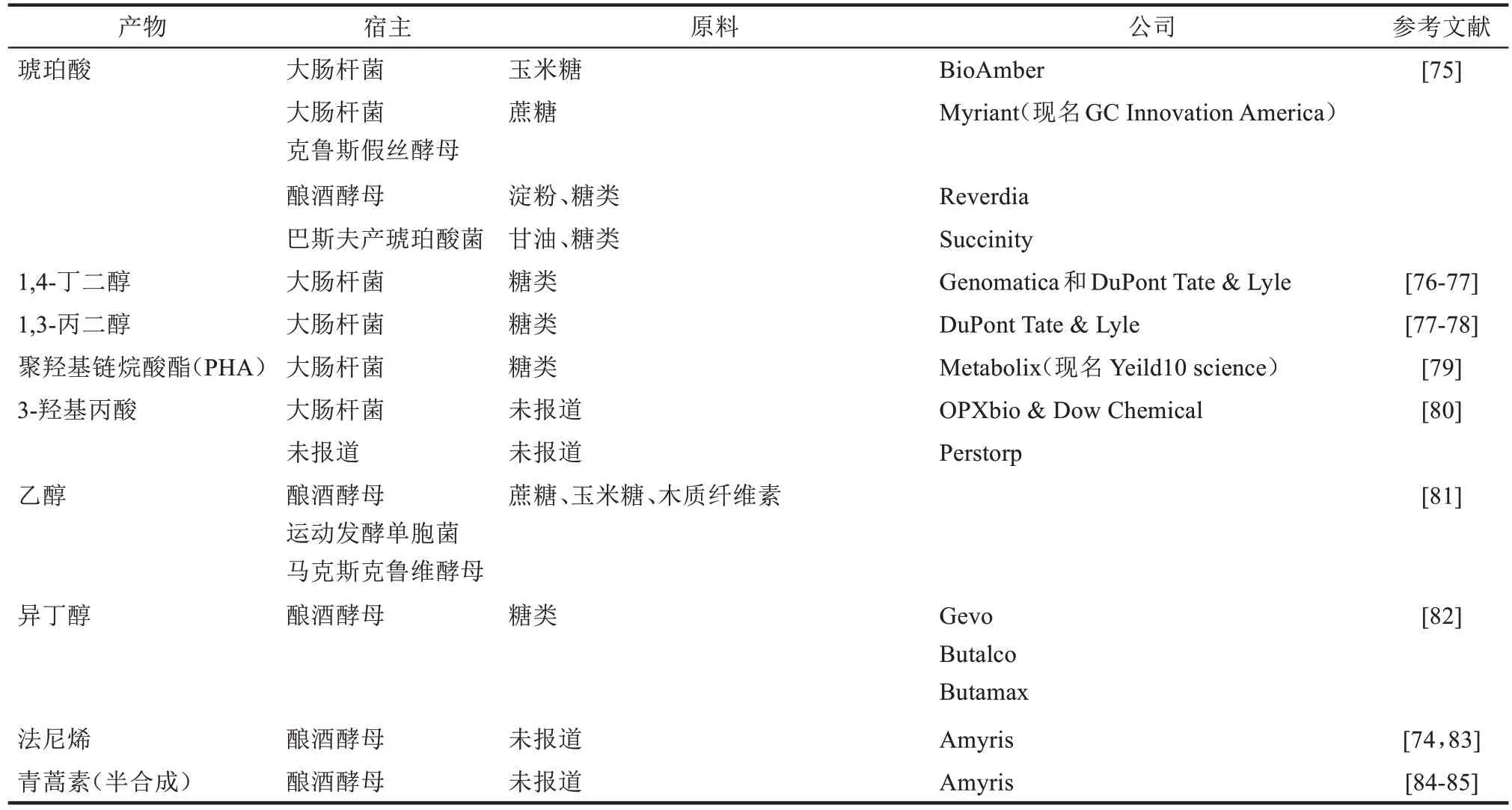
表4 代谢工程指导的经典设计策略的商业化应用案例Tab.4 Commercial application of classic design strategies guided by metabolic engineering
为了更为高效地设计和构建MCFs,必须快速高效地挖掘基因组范围内与目标表型相关联的未知基因及其位点。近年来,高通量基因编辑技术和表型筛选技术的发展有可能为未来MCFs的构建范式带来革命性的变革。在基因型方面,首先利用基因型高通量构建技术实现模式微生物全基因组范围内基因的高效编辑、突变和表达水平调节;在表型方面,利用表型高通量表征及筛选技术实现目标表型的高效表征和筛选;结合基因型高通量构建和表型表征/筛选获得的大容量混合文库样本,利用新一代测序和数据统计分析手段,建立特定表型关联基因及其位点GPA 数据集;最后,利用大规模GPA 数据集,结合深度学习等数据科学手段,基于数据科学手段从全基因组范围深度挖掘传统分子生物学手段无法发现的未知关联基因及其位点,将有可能从数据(学习)的角度绕开现有理性设计的知识局限性,进一步指导高效MCFs 的构建,为提高MCFs 设计和创建效率提供全新的研究范式。以下将对这一研究范式涉及的不同技术环节进行概要介绍。
3.1 基因组高通量编辑技术
微生物的基因组改造(敲入、敲除和引入突变)最早是基于同源重组技术实现的。最为广泛应用的Red/ET 同源重组技术利用单链DNA(singlestrand DNA,ssDNA)的重组实现基因组的改造,然而由于重组效率的限制,这一方法仅限于对单个基因进行串行操作,通量低,远不能满足全基因组范围大量基因编辑的需求。2009 年,George M.Church 团队提出了多重自动化基因组工程(multiplex automated genome engineering,MAGE)技术[89],通过对Red/ET 同源重组的参数进行优化并提出一套自动化装置,将单次ssDNA 重组效率提高到约30%。利用这一技术同时将靶向基因组上多个位点的ssDNA 文库转入细胞中,能够实现基因型的大规模并行改造。2010 年,Ryan T. Gill 团队提出了可追踪多重重组技术(trackable multiplex recombineering,TRMR)[90],通过在引入的 DNA中插入特异性标签(barcode),在实现基因组改造的同时,还可通过后续的标签测序反向追踪特定基因型。尽管MAGE 和TRMR 的出现极大地提高了微生物功能基因组学研究的通量,然而,高效同源重组工具只存在于少数模式微生物中,难以推广到更多具有应用价值的非模式微生物中。
近年来,CRⅠSPR 基因编辑技术的飞速发展为更为便捷的基因组编辑带来了新的机遇。CRⅠSPR-Cas 系统中的sgRNA 能够特异性靶向目标DNA 序列,根据不同需要引入Cas 蛋白(及其突变体或具有特定功能的融合蛋白),就能够实现目标基因的切割[91]、抑制[92]、激活[93]、编辑[94]和突变[95]。这类技术的可推广性远远高于前述的同源重组技术,目前已经被证明适用于包括古细菌在内的十余种重要的模式微生物[96-104]。由于sgRNA 只有20 bp 长,其本身就可以作为目标基因的特异性标签,实现混合文库中特定基因型的反向追踪。利用这一特性,结合DNA 合成和NGS 技术,能够实现sgRNA 文库的定制化合成和后续基于NGS 的低成本扩增子测序,从而极大地提高了功能基因组学研究的通量和便捷性。表5列举了近年来开发的不同类型基因组高通量编辑技术及其应用特性。
3.2 表型高通量表征/筛选技术
尽管基于CRⅠSPR-Cas 系统的高通量基因组编辑技术极大地提高了基因型的构建能力,在表型关联方面,现有研究主要以生长限制因素、耐药性、溶剂耐受性等为筛选条件,基于不同基因型微生物生长适应性(fitness)的差异,通过连续传代实现目的表型的筛选[29,86,90,108-110,114]。由于采用传统的摇瓶或孔板培养进行传代培养,将会耗费大量的时间和成本,限制了基因型-表型关联研究的效率。近年新发展起来的高通量微生物进化培养系统大幅提高了传代效率,例如,Wong 等[115]开发的eⅤOLⅤER设备能够同时大规模培养16种不同的微生物,其硬件、软件和湿件的高度模块化能够实现配置的快速更改,以适应更多高通量、自动化连续培养实验。本团队自主开发的基于液滴微流控技术的“微生物微液滴培养仪”(microdroplet microbial culture,MMC),结合微流控技术和光电传感与控制及自动化技术,可以实现微升级微生物液滴平行培养、生长曲线测定和适应性进化(通量102~104),对于典型的模式微生物如大肠杆菌、酿酒酵母、乳酸杆菌等,可实现稳定传代100代,大大提升了微生物底物利用能力、溶剂耐受性等表型样本获取的效率[116]。

表5 基因组高通量编辑技术Tab.5 High-throughput genotype construction technologies
除了基于耐受性的表型高通量筛选模型,近年来,还发展了一系列代谢物产量高通量筛选技术与装备。代谢物细胞传感器作为一类重要的合成生物学工具,能够通过特定的转录因子、核糖体开关等识别元件响应细胞内特定代谢物的浓度,并将其转化为荧光、抗逆生长等特定的输出信号,在代谢物浓度表型表征方面得到广泛应用。通过对天然识别元件进行鉴定或工程化改造,目前已经开发了不同的生物传感器[117-120]。代谢物细胞传感器与荧光激活细胞分选技术(FACS)相结合,可以将胞内代谢物浓度转换为易于检测的荧光信号,利用流式细胞仪可实现细胞的高通量表征及分选,目前已有诸多成功案例。然而,基于FACS的高通量代谢物检测技术仅局限于胞内代谢物浓度的检测,无法实现胞外代谢物的高通量检测。为了解决这些问题,研究者提出了基于液滴微流控的高通量表型筛选方法,将微生物包覆在一个液滴之中进行培养,再以液滴为单位进行胞外代谢物高产菌株的高通量筛选,表6列举了具有代表性的代谢物高通量表型表征/筛选技术,包括荧光激活液滴分选(FADS)、基于液滴的荧光激活细胞分选(Droplet-FACS)、基于凝胶微液滴的荧光激活细胞分选(Gel FACS)、拉曼活化液滴分选(RADS)等。
需要注意的是,目前的表型高通量表征/筛选技术仍然局限于特定时间点的单一表型快照(snapshot)。如果能够利用近年来快速发展的单细胞RNA 测序[128]、微生物原位高精度荧光显微定位追踪[129-131]等技术,使表型端能够输出高密度的时间序列信息,进一步结合合成基因线路[132],将输出表型扩展为单细胞水平异质性等更为重要和丰富的类型,将进一步推动表型高通量表征和筛选技术的发展,大幅度提升MCFs 的工程化能力。
3.3 基因型-表型关联技术
不同于阵列方法(array)对每个基因进行逐一敲除或过表达并进行单独研究的策略,基于混合文库筛选的方法通过高度平行化的实验,对基因组范围内的基因位点进行大规模的扰动,并检测特定筛选条件下的表型变化,能够实现功能基因的快速筛选。以基于CRⅠSPR 体系的混合文库筛选方法为例,其基本原理是利用定制化设计和合成的sgRNA 文库产生混合基因型文库,并在特定的筛选条件下进行筛选,随后利用NGS 技术对sgRNA 进行扩增子测序,根据测序结果分析筛选前后sgRNA 丰度的变化,从而获得特定基因型与筛选条件的关系,进而绘制全基因组规模的GPA图谱,实现未知功能基因位点的深度挖掘。

表6 微生物代谢物高通量表征/筛选技术Tab.6 High-throughput selection/screening technologies in single-cell level
目前,这一策略已经被逐渐引入到代谢工程领域并用于菌株工程化改造,其中的典型代表团队是美国科罗拉多大学的Ryan T.Gill团队。例如,他们利用CREATE 技术在大肠杆菌中针对19 个基因构建了库容为16 300 的混合文库,以赖氨酸类似物的竞争性抑制作为致死筛选条件,对赖氨酸代谢相关的基因进行了深度扫描,确定了lysP、argT和cadB等与类似物竞争性相关的基因[133]。利用类似的策略,该团队先后实现了异丙醇[134]、3-羟基丙酸[135]、苯乙烯[136]等高产菌株的构建。本团队利用CRⅠSPRi 技术首次在大肠杆菌中构建了靶向全基因组范围的sgRNA 文库,结合混合文库筛选的策略CRⅠSPRi-seq,实现了大肠杆菌全基因组(4000 余个基因)范围内必需基因、代谢网络结构、与糠醛和异丁醇耐受性相关基因的快速筛选和鉴定[29]。对于CREATE 技术,其突变位点挖掘的分辨率在碱基层次,由于数据密度极高,受限于建库和测序成本,目前只能实现部分基因的突变位点关联分析,相比之下,本团队开发的CRⅠSPRi-seq 方法分辨率在单基因层次,能够实现全基因组范围功能基因的高效挖掘。
3.4 全基因组规模定制工程中GPA数据的利用
如前所述,快速发展的NGS 技术和基因型-表型关联技术使得目前能够以较低的成本快速获得大量GPA数据。基于大规模并行实验表征获取的GPA数据集(103以上)为机器学习提供了高质量的注释样本,如何利用数据科学手段学习基因型-表型数据背后隐藏的“机制”或“规律”成为研究者日益关注的重要方向。目前,计算生物学领域已经发展了多种对不同层次的组学数据进行分析、挖掘和学习的算法[137]。结合丰富的组学数据,机器学习算法被成功应用于通路动力学的预测[138]、酵母5′非翻译区调控序列的研究[139]等。如果能够充分利用大规模GPA数据集,依托数据科学手段从全基因组范围深度挖掘传统分子生物学手段无法发现的大量的未知关联基因及其位点,将有可能从数据(学习)的角度绕开目前理性设计的知识瓶颈,实现定制化MCFs的快速、高效创制。
目前,机器学习手段在GPA数据的学习和利用方面还刚刚起步,相关研究还比较少。本团队尝试了小规模GPA数据在酵母MCFs构建中的应用。为了实现酿酒酵母中异源代谢途径表达水平的快速优化,我们提出了一种与酵母体内生物标准元件组装策略相结合的机器学习工作流程(machine-learning workflow in conjunction with YeastFab assembly,MiYA)[140],应用人工神经网络(artificial neural network,ANN)模型,以组合空间中2%~5%的数据为训练集,成功预测了具有最优表达水平组合的β-
胡萝卜素和紫色杆菌素合成途径。类似地,机器学习策略已经被用于指导构建生产特定风味物质的啤酒酵母[141]和色氨酸高产酿酒酵母菌株[142],展现出重要的应用潜力。此外,结合高通量基因组编辑和筛选技术,本团队还报道了迄今为止在大肠杆菌中规模最大的sgRNA活性数据集,并利用机器学习手段建立了sgRNA序列影响活性的数学模型,可用于预测并优化CRⅠSPR-Cas系统在大肠杆菌中的工作性能[143]。
4 展 望
MCFs 的设计和构建最终目标都是实际应用,市场需求快速多样的变化对MCFs构建效率和性能的提升提出严苛的要求。如图3所示,早期的诱变育种采取非理性手段进行MCFs 改造,是典型的“以时间(人力)换水平”的策略。随着生物学知识的积累,经典代谢工程的发展使得对生物代谢网络进行理性/半理性设计成为可能,以DBTL 循环为基本流程,MCFs 改造效率得到显著提升。系统代谢工程的建立进一步使得研究者能够结合组学和生物信息学手段获取生物学知识,从系统层次进行MCFs 的设计,进一步加快MCFs 的构建效率。然而,由于微生物代谢网络结构及其调控机制的复杂性和“生命暗物质”的广泛存在,基于上述策略进行MCFs设计和构建的过程仍然需要耗费大量的时间和精力,且由于搜索的基因组空间有限,难以满足工业生产不断增长的表型“高地”需求。随着高通量研究技术的发展,由数据驱动的全基因组规模定制工程化有望克服这些难题,通过将高通量技术在全基因组范围基因型空间的挖掘与改造相结合,有望以更低的开发成本、更短的研发周期获得生产效率更为高效、生产性能更加优越的下一代定制化MCFs。
全基因组规模定制工程化MCFs作为全新的研究领域和范式,目前还处于萌芽阶段。为了实现MCFs的定制化设计与构建,亟需解决如下两个重要问题:
第一,高通量高质量GPA 数据的产生。基因型端的基因组编辑技术、测序技术发展较为成熟,而在表型端,由于测试目标和测试条件的复杂性,目前还无法弥补与大规模基因型样本之间的数据鸿沟。自动化技术和高通量新型表型表征技术的引入是解决这一问题的重要发展方向。近年来,国内外基于自动化机器人的BioFoundry 平台建设方兴未艾,单细胞质谱、单细胞RNA 测序等技术也在迅速发展,这些都将为跨越基因型-表型数据鸿沟,实现高通量高质量GPA数据供给提供重要机遇。
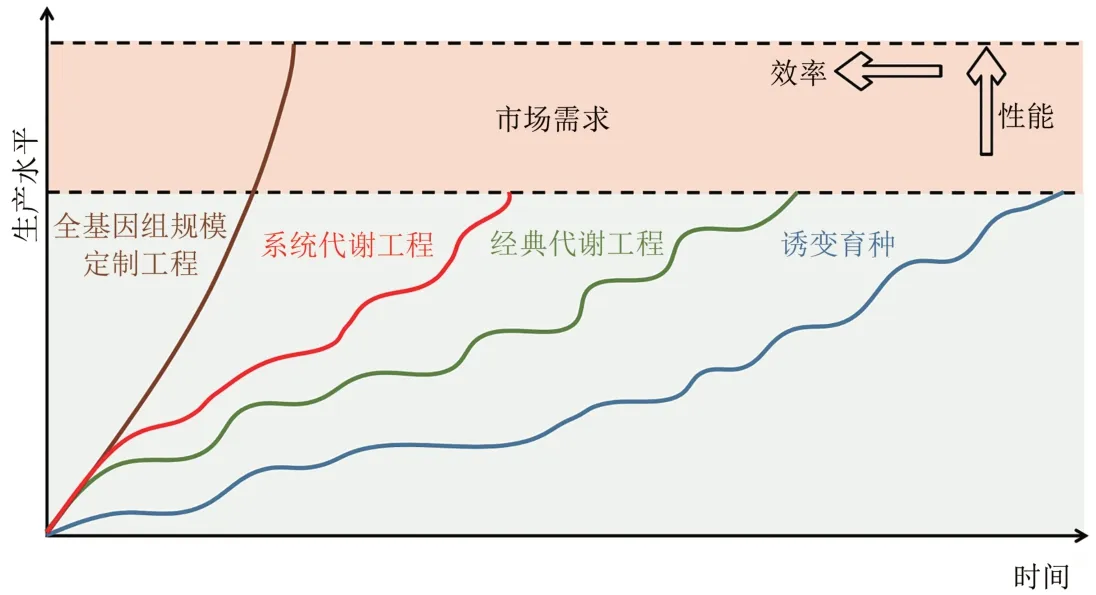
图3 微生物细胞工厂设计和构建策略效率以及性能对比Fig.3 Comparison of MCFs construction efficiency and performance in different stages
第二,数据管理和学习。高通量和自动化技术在极大地提升数据获取能力的同时将不可避免地带来数据爆炸,这为数据存储、数据标准化和数据共享带来了挑战。为了实现不同数据的共享和集成,迫切需要建立可查找、可访问、可相互操作和可重复使用的工程生物学数据库。此外,在如何利用GPA数据集指导定制化MCFs的设计和构建方面,亟需系统创建性能更优的生物数据分析、挖掘、学习算法和工具,以充分发挥大规模数据的效力。近年来基于卷积神经网络的深度学习算法、基于生成对抗网络的生成学习算法都被逐渐应用到生物系统领域,需要加快这些算法在GPA数据集学习和利用方面的探索和应用。
生物产业是我国国民经济的重要支柱,菌种是生物产业命脉所系,然而,我国菌种长期面临国外技术垄断、专利封锁的困境,近年更是频繁出现生物发酵产业核心菌种的国际知识产权纠纷。我们期待,如果能够抓住全基因组规模定制工程化发展机遇,提升我国MCFs创新能力,将有可能在未来日益激烈的国际竞争中占领菌种创制的战略制高点,实现我国生物产业的跨越式发展。
符号说明
BNICE——生化网络集成计算浏览器(biochemical net⁃work integrated computational explorer)
COBRA——基于约束的重构与分析(constraint-based reconstruction and analysis)
CREATE——基于CRISPR 的可追踪基因组工程(CRIS⁃PR-enabled trackable genome engineering)
CRISPR——规律成簇的间隔短回文重复(clustered reg⁃ularly interspaced short palindromic repeats)
CRISPRa——CRISPR激活(CRISPR activation)
CRISPRi——CRISPR干扰(CRISPR interference)
FACS——荧光激活细胞分选(fluorescence-activated cell sorting)
FADS——荧光激活液滴分选(fluorescence-activated drop sorting)
FBA——通量平衡分析(flux balance analysis)
GPA——基因型-表型关联(genotype-phenotype asso⁃ciations)
GSMM——全基因组代谢模型(genome-scale metabolic model)
GSMR——基因组规模代谢重构(genome-scale meta⁃bolic reconstruction)
MAGE——多重自动化基因组工程(multiplex automat⁃ed genome engineering)
MCA——代谢控制分析(metabolic control analysis)
MFA——代谢通量分析(metabolic flux analysis)
MMC——液滴微流控培养(microdroplet microbial culture)
MOMA——最小化代谢调节(minimization of the meta⁃bolic adjustment)
MPA——代谢途径分析(metabolic pathway analysis)
NET——网络嵌入的热力学分析(network-embedded thermodynamic analysis)
NGS——二代测序(next generation sequencing)
ODE——常微分方程(ordinary differential equation)
PHA——聚羟基链烷酸酯(polyhydroxyalkanoate)
RADS——拉曼活化液滴分选(raman-activated droplet sorting)
ROOM——开/关最小化调节(regulatory on/off minimi⁃zation)
sgRNA——向导RNA(single-guide RNA)
ssDNA——单链DNA(single-strand DNA)
TAM——靶向AID 介导的诱变(targeted AID-mediated mutagenesis)
TMFA——基于热力学的代谢通量分析(thermodynamicsbased metabolic flux analysis)
TRMR——可追踪多重重组(trackable multiplex recom⁃bineering)
YOGE——酵母寡核苷酸介导的基因组工程(yeast oligomediated genome engineering)
猜你喜欢
传染病信息(2022年4期)2022-11-23
河南医学研究(2022年18期)2022-09-30
西部医学(2022年9期)2022-09-26
中国典型病例大全(2022年11期)2022-05-13
河北果树(2021年4期)2021-12-02
天津医科大学学报(2021年1期)2021-01-26
透析与人工器官(2020年1期)2020-11-16
中华养生保健(2020年7期)2020-11-16
医药前沿(2020年20期)2020-11-10
中国现代中药(2019年5期)2019-07-03
