
一个肌张力障碍-帕金森综合征的家系调查及致病基因研究
2021-01-05 02:44高晨雨黄婷陈睿刘春梅张颖冬田有勇
临床神经病学杂志 2020年6期
高晨雨,黄婷,陈睿,刘春梅,张颖冬,田有勇
肌张力障碍-帕金森综合征(DP)是同时出现肌张力障碍和帕金森样表现的疾病的统称[1],其临床特点为同时出现肌张力障碍(定义为持续或间歇性的不自主肌肉收缩所导致的异常重复运动和/或异常姿势的运动障碍)以及震颤、肌强直、运动迟缓、步态和平衡障碍的帕金森综合征表现。从病因角度通常将DP分为散发性和遗传性两大类,散发性的有合并肌张力障碍的帕金森病、进行性核上性麻痹、多系统萎缩、皮质基底节变性病等;遗传性的如多巴反应性肌张力障碍、快发病性DP、X-连锁DP、少年型帕金森综合征、Wilson病、脊髓小脑共济失调等[2]。国内外对同时出现肌张力障碍及帕金森综合征症状的疾病的研究文献较少,其中的遗传性疾病已经确定了多种致病基因,包括常染色体显性遗传基因(ATP1A3)、常染色体隐性遗传基因(PARK2,ATP7B等)和X-连锁遗传基因(TAF1)[1-3]。本研究报告一个DP家系,收集了先证者相关临床资料并进行了家系调查,对该家系的致病基因进行了检测分析。
1 对象与方法
1.1 对象 本家系为江苏省南京市江宁区人,汉族。家系调查追溯三代共9人,2人有临床症状,除先证者外,另有一名症状类似患者(先证者姑母)已去世(图1);均表现有肌张力障碍和帕金森综合征。取得知情同意后,采集先证者一家三代,包括先证者、父亲、母亲、妹妹(2人)、弟弟(1人)、女儿(1人)及丈夫共8人的全血标本,经典方法提取基因组DNA。对先证者进行了病史体征采集及在未服药状态下进行统一帕金森病评定量表(UPDRS)、统一Wilson病评分量表(UWDRS)、Wilson病全球评估量表(GAS for WD)以及MMSE、蒙特利尔认知评估量表(MoCA)评估。常规实验室血液生化、甲状腺功能及铜蓝蛋白等检测。
1.2 方法
1.2.1 角膜裂隙灯、肝脏超声、MR影像及PET成像检查 对先证者行角膜裂隙灯检查拍照、肝脏超声检查排查Wilson病;行头颅、颈段和腰段脊髓3.0 T MRI扫描排除头颅、颈段及腰段的脊髓病变对头面及下肢运动异常的影响;行头颅18F-dopa PET/MRI黑质、基底节区多巴显像明确中枢多巴胺能神经元的受累情况。
1.2.2 基因分析 采用第二代测序技术和Sanger测序技术对先证者进行帕金森病相关48个已报道基因的外显子编码区测序(北京圣谷检验)。在此基础上,采用目标区域捕获高通量测序技术,对先证者行全外显子测序,家系7人行Sanger测序验证(杭州祥音生物)。所有可疑突变位点使用美国医学遗传学与基因组学学会(ACMG)遗传变异分类标准与指南[4]进行分类。SIFT软件和Polyphen-2软件进行蛋白功能及保守性预测。
2 结 果
2.1 先证者资料 先证者(图1Ⅱ-2),女,48岁,进行性行走困难10年。2009年5月(38岁)起出现左小腿痉挛性疼痛,并感行走不便,X线检查未见异常,诊断:帕金森病,予“盐酸金刚烷胺片”治疗(200 mg/d)治疗,效果不明显。后进行性加重,感左下肢僵硬、伸展受限,出现拖步。2013年觉行动缓慢较前加重,次年再就诊,予“美多芭”治疗(冲击试验:第1 d,125 mg/d;第2 d,187.5 mg/d;第3 d,250 mg/d;第4 d,312.5 mg/d;第5 d,375mg/d;症状改善不超过30%,维持375 mg/d治疗),仍无效。同年行骨髓穿刺无异常,期间虽活动不便,仍能坚持保洁工作。2018年僵硬、行走困难加重明显,并伴上肢轻度震颤,间断出现不自主眨眼(紧张时加重)及睡眠状态下磨牙。外院头颅CT检查无异常,中脑超声检查显示左侧中脑黑质强回声。予“美多芭”(250~375 mg/d)、“泰舒达”(50 mg/d)治疗效果欠佳,出现阴道不规则流血(此时已绝经2年),被迫停用。后反复多次用“美多芭”及“泰舒达”治疗均无明显效果,且出现阴道不规则流血,停药后消失。治疗过程中发现“苯海索片”(2 mg/d)及“氯硝西泮”(1~2 mg/d)可显著改善部分症状,包括拖步、不自主眨眼及磨牙。2019年11月收住院。患者无吸烟饮酒史,否认药物、毒物及特殊环境暴露史。父亲(图1Ⅰ-2)2019年罹患胃癌,姑母(图1Ⅰ-1)有震颤、动作不稳等类似症状,已去世,其他家族成员身体健康。查体:体型正常,内科查体无明显异常。神经专科查体:面部表情僵硬,瞬目频繁,下颌不自主咬合动作。言语低沉带爆破音。上肢对称性静止性震颤,四肢肌力正常,肌张力增高,左侧更甚。两侧深浅感觉正常。起步缓慢,启动后左下肢拖曳、步态前冲。四肢腱反射(),右侧Babinski征可疑阳性。未服药状态下:UPDRS-Ⅰ评分10分,UPDRS-Ⅱ评分14分,UPDRS-Ⅲ评分46分,GAS for WD评分14分,UWDRS-Ⅰ评分54分,UWDRS-Ⅱ评分2分,UWDRS-Ⅲ评分1分,MMSE评分30分, MoCA评分26分。实验室检查:血常规、生化、甲状腺功能、凝血功能均正常;血清铜蓝蛋白0.28 g/L。
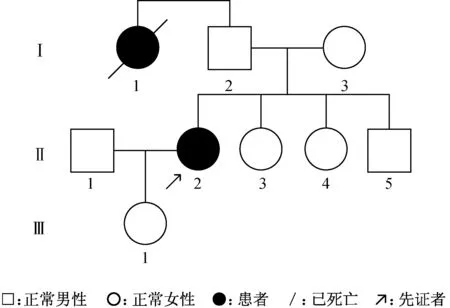
图1 DP家系图。 三代共9人,出现临床症状2例,1例死亡
2.2 裂隙灯眼角膜数码照相 先证者裂隙灯下双眼未见角膜K-F环(图2)。
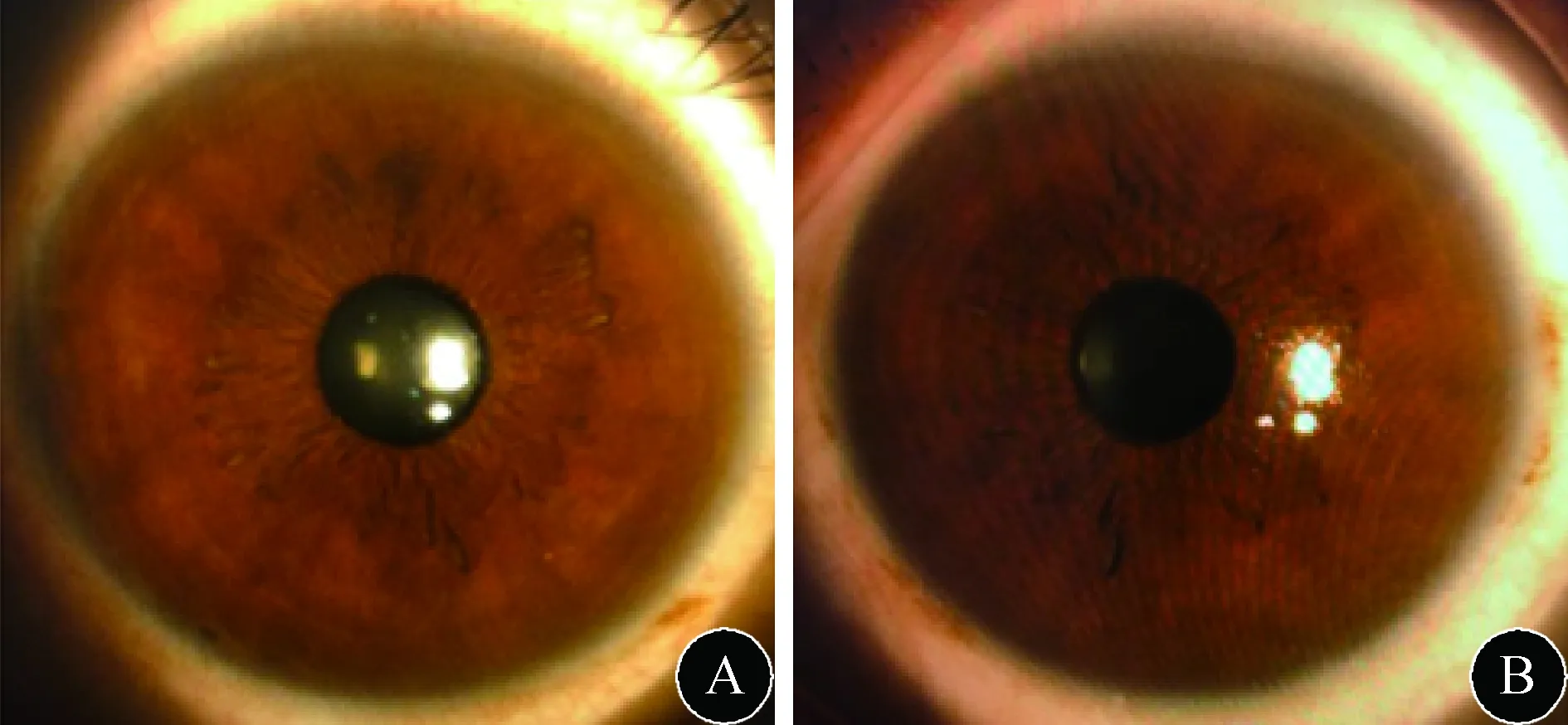
图2 裂隙灯眼角膜数码照相。 A:右眼;B:左眼,均未见K-F环
2.3 超声 先证者肝脏超声未见明显异常(图3)。
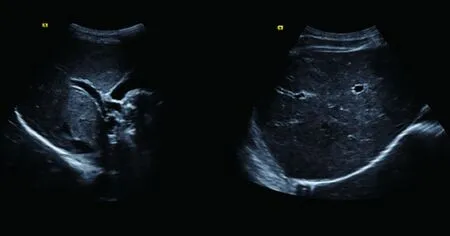
图3 肝脏超声。肝脏形态大小未见明显异常,实质回声均匀,包膜光滑,肝内管状结构清晰
2.4 MRI检查 先证者头颅T1、T2、Flair-MRI无明显异常;颈、腰椎MRI仅颈腰椎体轻度退变。脊髓MRI无异常(图4)。
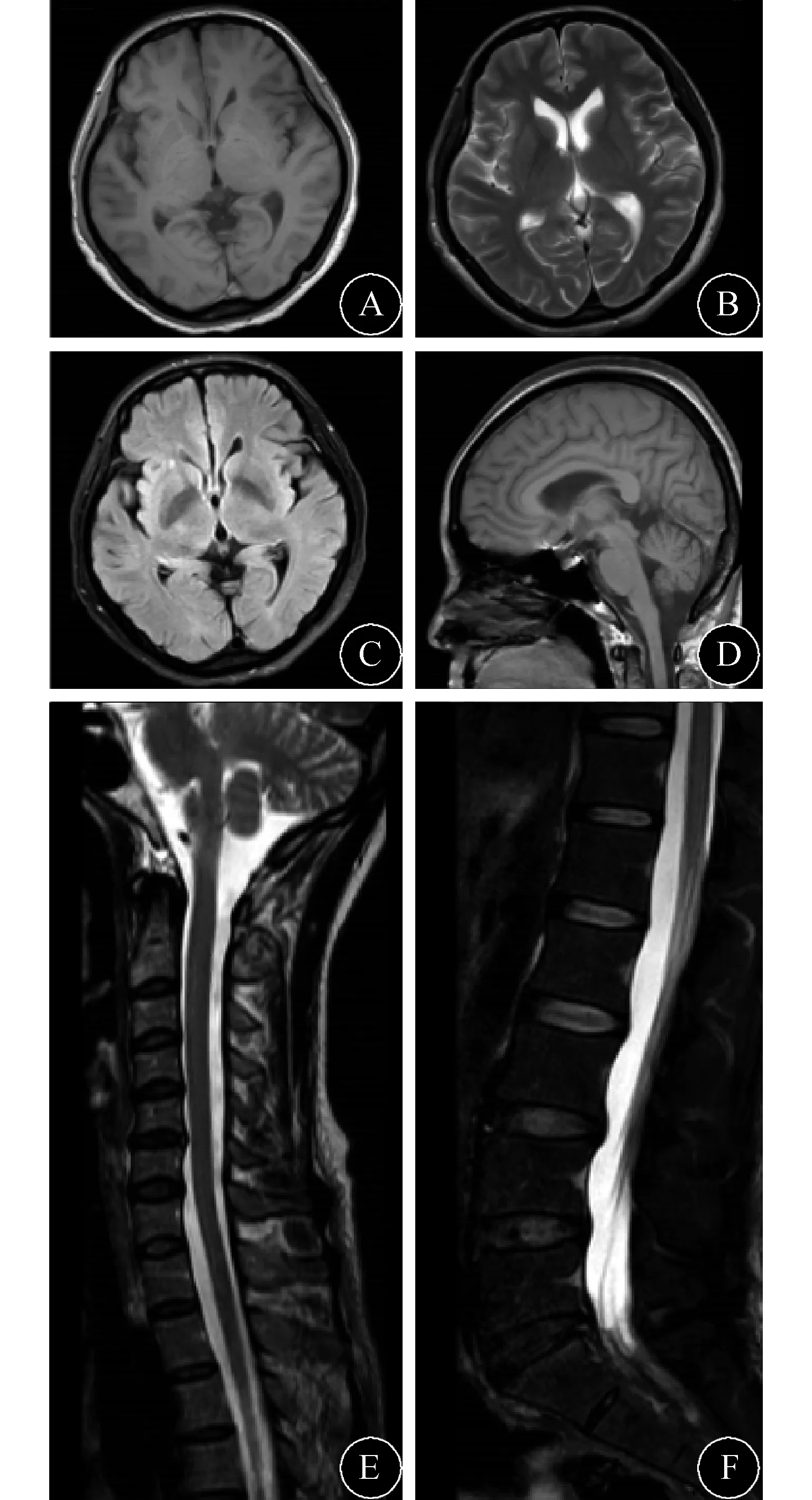
图4 MRI结果。A~D:头颅MRI双侧脑干、基底节无明显病变,无明显中脑、脑干萎缩;E:颈椎MRI仅见C3~C7轻度椎间盘脱出;F:腰椎MRI见L4~S1椎间盘突出或膨出,未累及脊髓
2.5 PET/MR成像 先证者脑18F-dopa PET/MRI显像示双侧纹状体和黑质显影清晰,双侧基本对称,双侧豆状核多巴胺神经元放射性摄取略减低(图5)。
2.6 基因突变分析 采用NGS+Sanger测序验证技术,对先证者进行帕金森病相关48个已报道基因的外显子编码区进行检测,结果显示在先证者(Ⅱ-2)ATP7B基因外显子18存在c.3792G>C (p.M1264I) 的杂合错义突变(图6),ACMG分类为临床意义未明(PM2+PP3),SIFT软件预测该突变为有害的(deleterious),Polyphen-2软件预测为可能致病的(probably damaging)。
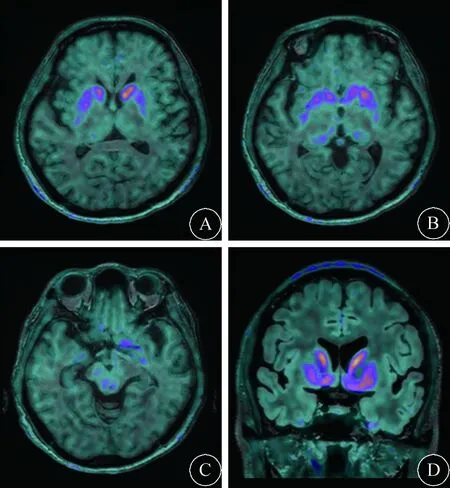
图5 18F-dopa PET/MRI结果。双侧豆状核多巴胺神经元放射性摄取略减低
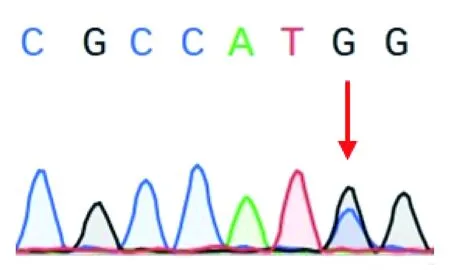
图6 ATP7B c.3792G>C(p.M1264T) Sanger测序图
采用目标区域俘获高通量测序技术,对先证者血样行进一步的全外显子测序及家系Sanger测序验证。结果发现,先证者(Ⅱ-1)及另外3人(Ⅰ-2、Ⅱ-4、Ⅱ-5)携带父源RYR1基因36号外显子c.5972C>T(p.T1911I) 的杂合错义突变(图7A),ACMG分类为临床意义未明(PM2+PP2+PP3),SIFT软件预测该突变为影响蛋白功能的(affect protein function),Polyphen-2软件预测为可能致病的(probably damaging)。其他家庭成员(Ⅰ-3、Ⅱ-1、Ⅱ-3、Ⅲ-1)均为野生型(图7B)。
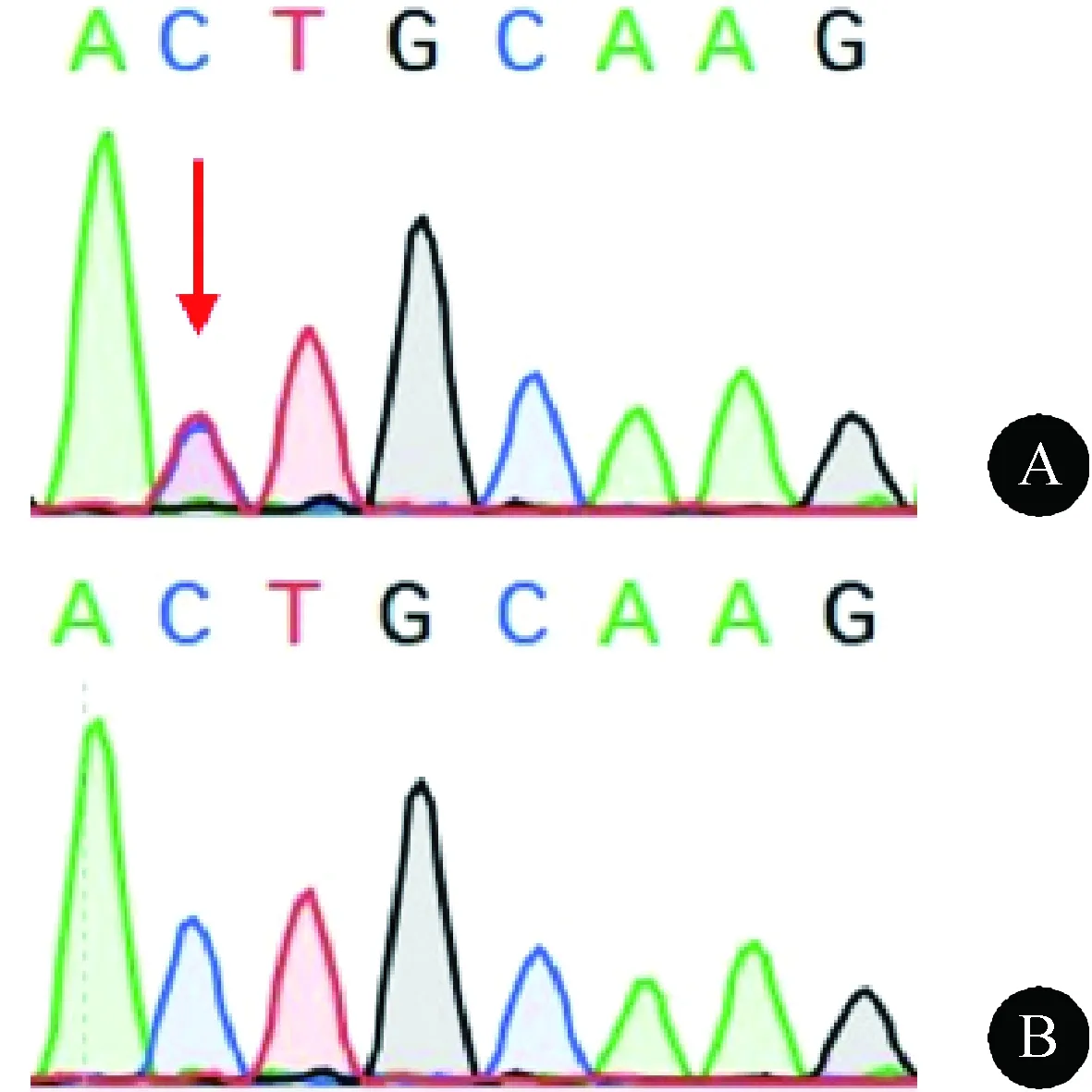
图7 RYR1 c.5972C>T (p.T1991I)Sanger测序图。A:突变型;B:野生型
除上述突变外,全外显子测序还检测到先证者携带母源的SETX基因10号外显子c.2439A>C(p.E813D)杂合突变(图8A)和GCDH基因10号外显子c.1060G>A(p.G354S)杂合突变(图9A),先证者二妹及女儿(Ⅱ-4、Ⅲ-1)亦携带GCDH基因的杂合突变,其他家庭成员(Ⅰ-2、Ⅱ-1、Ⅱ-3、Ⅱ-5)的上述基因均为野生型(图8B、图9B)。上述突变致病意义均未明确。
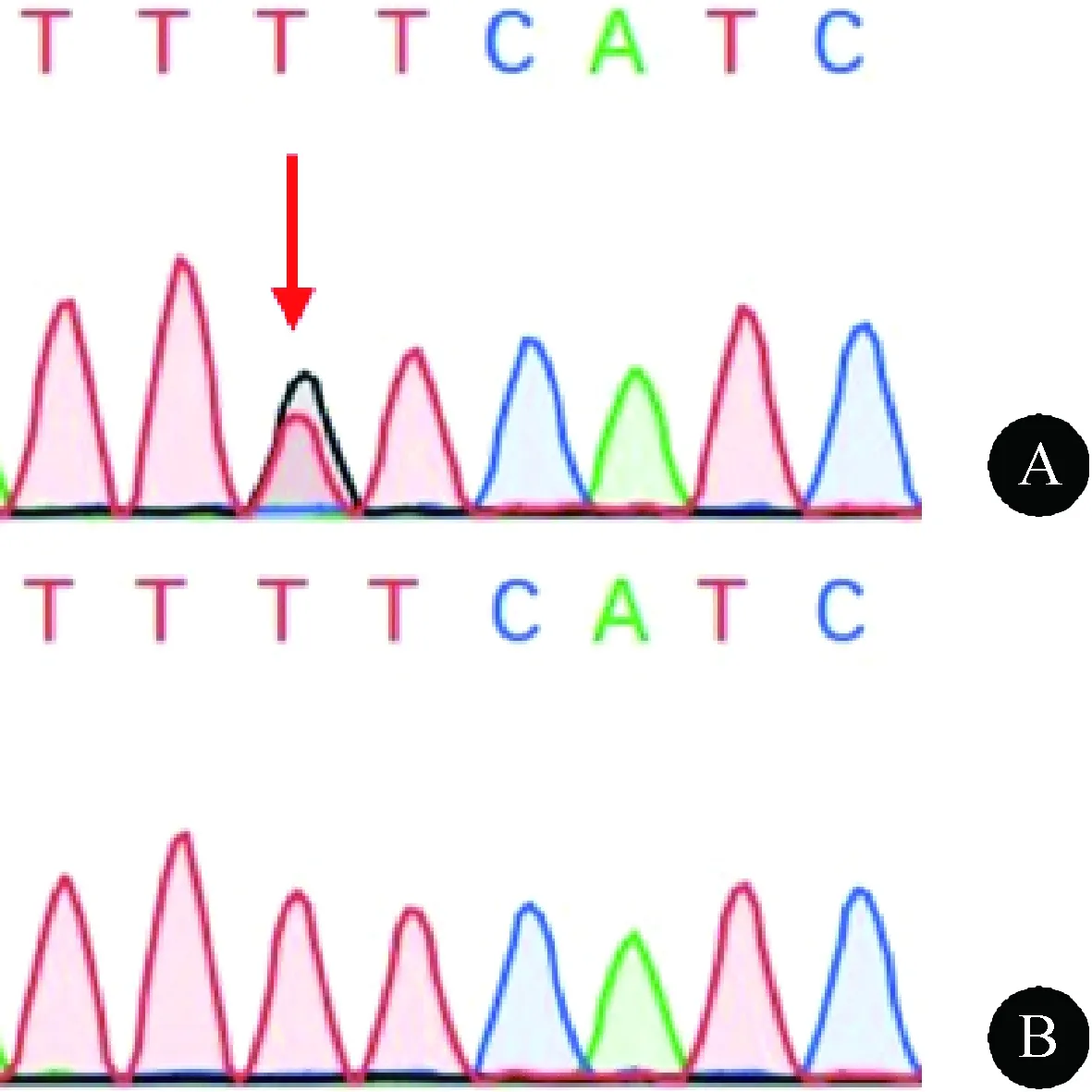
图8 SETX c.2439A>C(p.E813D) Sanger测序图。A:突变型;B:野生型
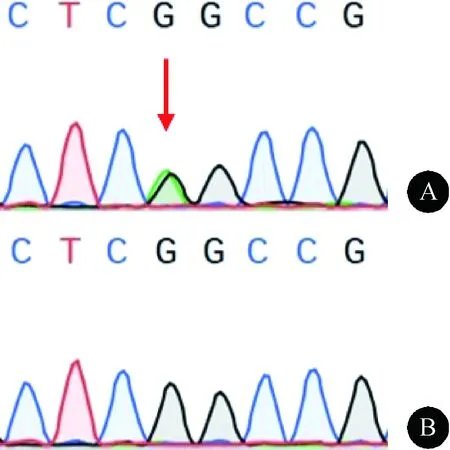
图9 GCDH c.1060G>A(p.G354S) Sanger测序图。A:突变型;B:野生型
3 讨 论
先证者中年(38岁)起病,病史10年,以单侧下肢痉挛性疼痛和行走不灵活为首发症状,进展缓慢,逐步出现动作慢,肢体僵硬,轻度震颤,肌张力高等表现,同时伴不自主眨眼、口部咬合动作及夜间磨牙和下肢行走拖拽步态。既有帕金森综合征的症状,又兼有肌张力障碍的表现,考虑到以肌张力不全表现首发且症状突出,临床诊断为DP。根据相关神经功能评分,症状以锥体外系运动功能受损为主,但对多巴胺能药物治疗作用不敏感,可排除帕金森病。经过对角膜、肝脏、脑影像学及铜蓝蛋白检查,临床排除Wilson病。头颅PET/MRI多巴显像检查示多巴胺系统有受损,提示先证者存在帕金森综合征相关疾病可能。先证者没有认知功能受损,植物神经损伤症状也不突出,无眼球活动障碍,结合头颅MRI结果,路易体痴呆、额颞叶变性、进行性核上性麻痹、多系统萎缩等可能出现DP的常见疾病均无诊断依据。脊髓MRI也排除了脊髓损伤之可能。因此,本患者病因仍不明确;多巴胺能药物治疗过程中出现阴道出血可能与药物治疗相关,但未见类似报告。
病史询问中,先证者提供其亲属姑母罹患相似临床症状,使得考虑到该案例是否为某种遗传性疾病可能。首先针对先证者进行了帕金森病相关基因的靶向测序,结果仅发现ATP7B基因外显子18存在c.3792G>C (p.M1264I) 的杂合错义突变。经查证,该变异未收入HGDM数据库,ESP数据库、千人数据库及ExAC数据库的正常人群对照中均无突变频率数据,亦没有检索到相关文献报道。Dong等[5]曾报道,在Wilson病患者中检测到ATP7B基因c.3791 T>C (p.M1264T)的突变,并归类为疑似致病突变,该突变HGDM数据库ID为CM164061,变异等级为DM(致病突变)。ATP7B基因变异导致的Wilson病呈常染色体隐性遗传,单个杂合突变不足以引起疾病发生[6]。因此,用该基因的单个杂合突变不能解释先证者的遗传学病因。
为排查先证者可能的遗传学病因,我们在进行靶向测序结果失败的情况下,又采集了先证者家系血样,进一步采用目标区域俘获高通量测序方法进行了全外显子测序,发现先证者还携带来自父系的RYR1基因36号外显子c.5972C>T (p.T1991I)杂合错义突变,先证者妹妹、弟弟存在相同突变,但父亲、妹妹以及弟弟并无类似的临床症状。在GnomAD数据库中未见该基因突变频率报道,经SIFT软件预测该突变为影响蛋白功能, Polyphen-2软件预测为可能致病(probably damaging)。文献表明,该基因变异相关的疾病主要包括:(1)中央轴空病:多数为常染色体显性遗传,少数为常染色体隐性遗传,显性突变和隐性突变在RYR1基因上位置不同,显性遗传突变主要位于该基因100~102号外显子,而隐性遗传突变平均分布于该基因各个区域[7];(2)小核肌病伴眼外肌麻痹:为常染色体隐性遗传,累及线粒体功能[8]。以上两种已报道的疾病均表现为婴幼儿、儿童早期发病,以肌无力、肌萎缩、肌张力低下,伴或不伴眼外肌、呼吸肌、心肌受累和骨关节畸形等临床特点[8]。先证者所在家系遗传和临床特点和上述已报道疾病显然不符。因此,本家系出现的RYR1基因c.5972C>T杂合错义突变与本患者家系的致病证据需进一步研究。
以DP为主要表现的遗传性疾病还包括多巴反应性肌张力障碍以及快发病型DP等。前者至少已被发现3个致病基因(GCH-1、TH、DYT14),后者的致病基因为ATP1A3;然而先证者及其家系中均未发现上述两种类型DP的致病基因突变,且其发病年龄、起病形式以及对多巴胺能药物治疗的敏感性均与上述两种疾病不符[1,3,9]。
先证者全外显子测序中发现了另外两个遗传自母亲的杂合突变,即SETX基因外显子10的c.2439 A>C(p.E813D)杂合突变和GCDH基因外显子10的c.1060G>A(p.G354S)杂合突变,家系中仅有先证者及其母携带SETX基因c.2439A>C突变,而有包括先证者在内共4人携带GCDH基因c.1060G>A突变。但上述突变的携带者并无类似先证者的临床症状,且这两种基因相关的疾病(肌萎缩侧索硬化症4型、常染色体隐性脊髓小脑共济失调1型、戊二酸血症Ⅰ型)多为隐性遗传,其表型与本家系表现出的DP的临床表现差异较大,故难以合理解释本家系的病因。
综上所述,本研究报告的这类复杂临床综合征仍存在着诊断盲区,而采用的基因检测手段也存在一定的局限性。本研究采用的全外显子测序不能检测基因的调节区域和深度内含子区域的突变,不能反映染色体数目及结构异常以及某些 DNA 大片段的缺失、拷贝数变异和动态突变等特殊类型突变。此外,该家系中先证者姑妈临床资料有限,缺乏血标本进行测序,限制了家系的遗传学分析。本案例的具体遗传学病因还需要更多研究进行明确。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
临床输血与检验(2022年3期)2022-06-22
种子(2021年3期)2021-04-12
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
吉林林业科技(2018年6期)2018-11-21
中国生育健康杂志(2018年6期)2018-11-13
科技视界(2016年27期)2017-03-14
中学生理科应试(2016年7期)2016-05-14
听力学及言语疾病杂志(2015年5期)2015-12-24
