
团簇Co3 NiB2 的催化析氢活性
2021-01-03 10:05:12方志刚赵璐璐井润田
四川师范大学学报(自然科学版) 2021年1期
秦 渝, 方志刚, 赵璐璐, 井润田
(辽宁科技大学 化学工程学院,辽宁 鞍山114051)
目前,如何快速、有效地提高化学反应的速率和产率已成为各国研究的热点课题之一.催化剂作为一种既能提高化学反应速率又不改变化学反应最终结果的物质,不仅深受广大科研实践者的青睐,而且在理论研究领域也占有十分重要的地位.近年来,在Co -Ni 体系中掺杂各种金属或非金属元素以获得性能优化的研究已经有了很大的突破,其中在非晶态Co-Ni合金中掺杂B元素所形成的非晶态Co-Ni-B体系,已被实验证实其在催化活性方面具有非常好的效果.
由于一次能源的过度消耗所导致的自然资源缺乏与环境污染严重等问题,正迫使人们努力开发高效且绿色清洁的可替代能源.氢气作为一种新型清洁能源,在生理医学、军事工业、民用工业、电子工业及冶金工业等领域都占据着非同凡响的地位.此外,氢气因其原料丰富、产热量高、清洁环保、产物可循环利用等优点而备受世人关注.虽然硼氢化合物析氢和水电解析氢[1-3]是作为目前生产氢气的主要途径,但由于在析氢反应中,昂贵的技术成本以及贵金属催化剂的辅助使得该种方法在氢气析出方面进行大规模推广使用的愿望受到了阻碍.因此,对非贵金属催化剂的探索与研究便显得格外的重要与迫切.而非晶态合金Co-Ni-B作为非贵金属催化剂的一种,不仅在众多催化析氢实验中表现出了极佳的催化反应活性,而且在多次循环利用后仍可基本保持初始活性不被破坏[4-6].因此,为解决能源匮乏与环境污染问题,进一步开发出环保绿色的氢能源,对非晶态合金Co-Ni-B在析氢反应中的催化机制进行深入研究是非常有必要的.但截至目前,无论是在宏观实验还是在微观理论方面,对非晶态合金Co-Ni-B三元体系的相关研究甚少,且在已有的成果中,大多是对该体系催化性质的宏观实验研究,微观理论方面鲜少报道.鉴于此,本文根据文献[4]中Co、Ni、B 原子配比计算出的非晶态合金Co-Ni-B的最佳催化结构,通过对其稳定性和催化活性进行评估,并以已构建出的团簇Co3NiB2模型催化析氢反应为基础研究其前线轨道和内部电子结构,判断出团簇分子的反应活性强弱,从而设计出团簇Co3NiB2性能最优的催化剂构型.
1 理论模型和计算方法
1.1 设计理论模型迄今为止,用建立局部模型来模拟整体进行研究以探求和预测物质宏观的物理或化学性质是诸多科研工作者惯用的方法之一[7-9].为更好地探究出非晶态合金体系的催化性质,选择用团簇来模拟非晶态合金局部结构进行深入研究.依据文献[4]的实验过程及结果可知,当原子配比为n(Co)/n(Co+Ni)=0.85 时,Co -Ni -B 体系的催化性能最好,但由此计算得到的原子比为n(Co)∶n(Ni)=17∶ 3,所得金属原子数目较多,分析较为复杂.当n(Co)/n(Co +Ni)=0.75 时,Co-Ni -B三元体系的催化性能与n(Co)/n(Co+Ni)=0.85时相差无几,而由此计算得到n(Co)∶n(Ni)=3∶ 1,所得金属原子数目适中,更加便于进行理论模型的建立.此外,随着B 原子数目的增加,体系催化活性增强,故建立团簇Co3NiB2作为研究Co-Ni -B 体系的理论模型.基于拓扑学原理[10]对团簇Co3NiB2进行假想异构体的设计,共得到包括平面六边形、平面五边形(一个原子被其他5个原子包围)、五棱锥、四棱双锥、单帽四棱锥、单帽三棱双锥以及三棱柱构型在内的35种初始构型.
1.2 模型计算方法在B3LYP/Lan12dz 水平下,采用DFT[11-12]方法并利用启天M4390 计算机中的Gaussian 09 程序对团簇Co3NiB2所有初始构型在二、四重态下进行优化计算,对计算所得优化构型的B原子和Co、Ni原子分别采用Hay等[13]的Dunning/Huzinaga 双ξ 基组(9s,5p/3s,2p)与18 -eECP双ξ基组(3s,3p,3d/2s,2p,2d).
1.3 析氢反应机制以团簇Co3NiB2(文中用M表示)为主要模拟对象,其进行催化水析氢时的反应机制[14]如下:
团簇吸附氢原子(吸氢反应)
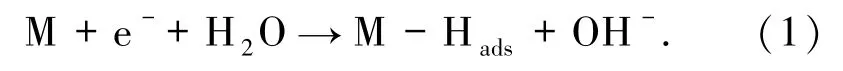
析出氢气反应(解吸反应,该反应可能存在以下2 种途径):
1)电化学解吸
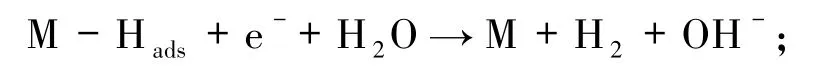
2)化学重组
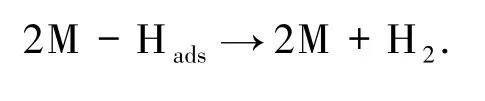
2 实验结果与讨论
2.1 团簇Co3NiB2 空间结构稳定性研究
2.1.1 团簇Co3NiB2的4 种优化构型 改变不同原子的相对位置,设计出35 种团簇Co3NiB2可能存在的初始构型,将团簇Co3NiB2在不同重态下优化后的各异构体中的相同构型及含虚频的不稳定构型排除之后,对剩余构型进行构型间的异构化转化研究,最终得到如图1 所示的4 种优化构型.其中,二、四重态各2 种.以五棱锥构型1(4)(其能量最低)为能量零点并计算出其余构型的相对能量,将剩余的优化构型分别按相对能量的高低依次排序(排序原则为由低到高).从图1 可以看到,团簇Co3NiB2能够稳定存在的4 种优化构型分别以五棱锥(1(4)与1(2))、单帽四棱锥(2(4))、单帽三棱双锥(2(2))这3 类空间结构存在.

图1 团簇Co3NiB2 的4 种优化构型Fig. 1 The 4 optimized configurations of cluster Co3NiB2
2.1.2 团簇Co3NiB2的稳定性分析 比较各优化构型的相对稳定性大小是研究团簇Co3NiB2催化活性的基础步骤之一,因而可更加清晰直观地分析结合能与吉布斯自由能变的相对关系,依据表1 中的热力学数据(能量EZPE、吉布斯自由能G、吉布斯自由能变ΔG以及结合能EBE)绘制出如图2 所示的团簇Co3NiB2的4 种优化构型EBE与ΔG的变化趋势图.
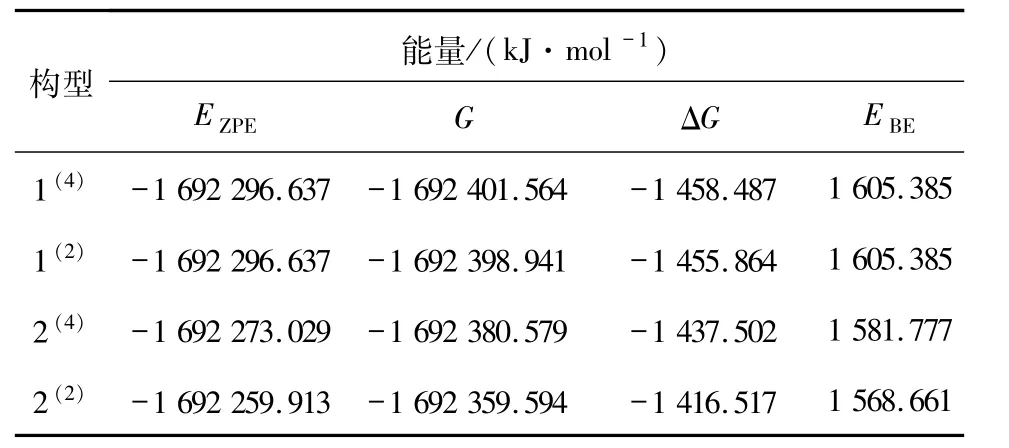
表1 团簇Co3NiB2的4 种优化构型的能量参数Tab. 1 Energy parameters of 4 optimized configurations of cluster Co3NiB2
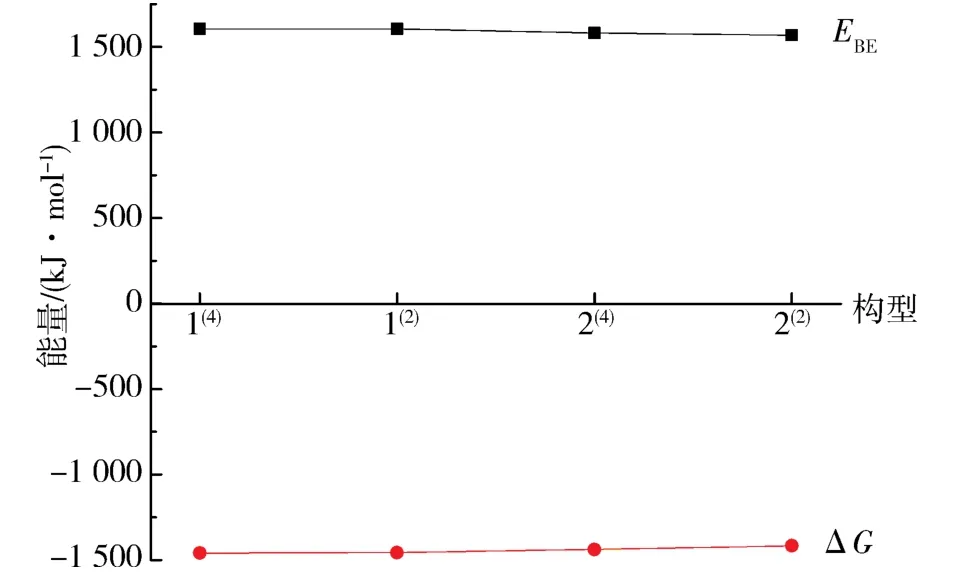
图2 团簇Co3NiB2 的4 种优化构型的结合能(EBE)和吉布斯自由能变(ΔG)Fig. 2 Binding energy(EBE)and Gibbs free energy change(ΔG)of 4 optimized configurations of cluster Co3NiB2
表1 中结合能(EBE)与吉布斯自由能变(△G)的计算公式分别为:

结合表1 与图2 可以看到:团簇Co3NiB2能稳定存在的4 种优化构型的EZPE、G、ΔG以及EBE的差异均较小.仔细观察图2 可以发现,构型2(2)的EBE与ΔG的绝对值较其余3 个构型有轻微的减少,相对应地,它的EZPE较其余3 个构型出现了较小程度的增加.当某一构型的EZPE越低、ΔG越小、EBE越大时,就意味着该构型的热力学稳定性越好.由此可知:在团簇Co3NiB2的4 种优化构型中,构型1(4)因其能量最低、吉布斯自由能变最小、结合能最大,而具有了最好的热力学稳定性;构型2(2)则与1(4)相反,其热力学稳定性最差.
2.2 团簇Co3NiB2 催化水析氢的吸氢反应
2.2.1 团簇Co3NiB2的4 种构型的HOMO图与水
分子的LUMO图 日本著名化学家Fukui[15]于20世纪50 年代提出的前线轨道理论,以其简单有效及化学概念明确的特点,在有机化学方面得到了广泛的应用,从而赢得了世界上化学工作者的密切关注.他认为,决定一个体系能否发生化学反应的关键是一同决定着分子的电子得失和转移能力的前线HOMO轨道与LUMO轨道,与此同时,能量最高的电子占据轨道HOMO 和能量最低的未占轨道LUMO也与团簇的反应活性密切相关.因此,有理由认为前线轨道在化学反应中起主导作用[16-17].团簇Co3NiB2在完成催化析氢机制的第一步反应(即团簇吸附氢原子)时,电子是从团簇Co3NiB2的HOMO轨道向水分子的LUMO轨道转移,从而实现了团簇原始优化构型与水分子中氢原子的吸附,并结合形成了(Co3NiB2)-H 的结构模型.为了更加直观的观察团簇Co3NiB2各优化构型HOMO 轨道与水分子LUMO轨道间电子的转移及相互作用情况,绘制出图3.不难发现,团簇Co3NiB2的二、四重态构型原子间均存在着α电子(自旋向上)与β 电子(自旋向下).为了更深入地探究出团簇HOMO轨道对水电解析氢反应的作用,对团簇二、四重态优化构型的HOMO轨道分α -HOMO与β -HOMO进行研究是十分必要的.从图3 中可以看到,在各优化构型的HOMO图及水分子LUMO图中均分布着颜色深浅不同的2 个阴影区域,其中被阴影覆盖的区域意味着电子在此处出现的概率密度较大,且颜色的深浅分别代表着轨道波函数相位的正负,浅色(图中为灰色)为正相,深色(图中为黑色)为负相.图中阴影部分电子云密度较大,即阴影部分为团簇Co3NiB2在参与催化析氢反应时更为活跃的部分.
观察图3 中H2O 的LUMO 轨道图发现,其外部全部为深色阴影,而浅色阴影仅在内部氧原子附近占据一小部分区域,说明水分子LUMO轨道中出现概率更大的是轨道波函数相位为负的电子.这就表明在与外来活化分子相互作用时,水分子更易接受其HOMO轨道中负相位的电子,从而达到分子轨道重叠成键的目的.接着再对团簇Co3NiB2各优化构型的HOMO 轨道图进行分析比较.为保证团簇Co3NiB2能更好地与水分子作用吸附上氢原子,只有当团簇HOMO轨道中轨道波函数相位为负的部分与水分子的LUMO轨道进行同号重叠,才更容易实现轨道间电子的转移与成键.整体观察发现,团簇二、四重态各优化构型α -HOMO图的深色面积均小于β -HOMO 图的深色面积,说明α -HOMO对团簇Co3NiB2构型HOMO 轨道中的负相位部分的贡献不及β -HOMO,即β -HOMO更容易与水分子的LUMO轨道重叠,从而使团簇Co3NiB2构型吸附上氢原子.另外,在β - HOMO 图中,除构型2(2)外其他构型的轨道波函数为负的部分几乎全在外围且其面积均大于轨道波函数为正的部分,说明团簇Co3NiB2优化构型的β -HOMO 轨道在参与反应给出电子时,会更有利于与LUMO轨道波函数相位为负的外来分子反应,展现出较好的反应活性.再对团簇各构型的α - HOMO 图进行分析发现,相比于与之对应的β -HOMO 图,其浅色阴影面积均较大且部分面积与相位为负的部分相互镶嵌,这说明团簇Co3NiB2各优化构型α -HOMO轨道在接近水分子的LUMO轨道时,轨道相位进行异号重叠的可能性更大,不能实现有效成键.综上所述,团簇Co3NiB2各优化构型的前线β -HOMO 轨道在催化水析氢的吸氢反应中起主导作用.

图3 团簇Co3NiB2 的4 种优化构型的HOMO轨道与水分子的LUMO轨道图Fig. 3 The HOMO orbital of 4 optimized configurations of cluster Co3NiB2 and the LUMO orbit of H2O
2.2.2 团簇Co3NiB2与水分子的前线轨道能级差由于图3中的阴影部分反映的是HOMO或LUMO轨道的电子离域空间,而非电子的具体离散情况.因此,仅通过比较团簇HOMO 轨道图与水分子LUMO轨道图进而得出团簇各优化构型与水分子反应的难易程度,存在一定的局限性.而根据Fukui提出的前线轨道理论[15]可知,参与化学反应分子间的前线轨道能级差越小,电子越容易在前线HOMO与LUMO 轨道间转移,故而反应越容易发生.由此可知,团簇Co3NiB2各优化构型的HOMO 轨道与水分子LUMO 轨道间的能级差值ΔE(ΔE=ELUMO-EHOMO)也是衡量团簇各优化构型进行催化反应难易程度的重要参数之一.与此同时,通过比较二者之间的能级差ΔE(见表2),还可以进一步分析出团簇Co3NiB2中催化析氢活性最佳的优化构型.
当反应物分子间的HOMO 与LUMO 轨道能级差小于579 kJ·mol-1时,反应易于发生,且能级差越小,反应越容易发生[14,18].这也就意味着要想使电子易于从团簇各构型的HOMO 轨道转移到水分子的LUMO轨道,从而完成团簇催化析氢反应机制第一步的前提条件就是使团簇Co3NiB2各优化构型的HOMO能级(EHOMO)与水分子LUMO能级(ELUMO)间的差值(ΔE)小于579 kJ·mol-1.由表2数据不难发现,团簇所有优化构型的ΔE均小于且远低于579 kJ·mol-1,说明在催化水析氢的吸氢反应过程中,团簇Co3NiB2各优化构型均能在一定程度上展现出较好的反应活性.再对团簇Co3NiB2各优化构型ΔE的数值大小进行具体的分析发现,在与水分子反应时,构型1(2)(ΔEmax= 529 kJ·mol-1)的HOMO轨道在给出电子与氢原子成键的过程中,其发生难度相比于其他3 个构型而言会稍有一定程度的增加;而构型2(4)(ΔEmin= 263 kJ·mol-1)则与之相反,在与水分子反应时其HOMO轨道中的电子最容易流入水分子的LUMO轨道中,从而使构型2(4)吸附上氢原子.此外,构型1(4)与2(2)两者的ΔE相近且较小,说明构型1(4)与2(2)在催化水析氢的吸氢反应中均能表现出较好的反应活性.

表2 团簇Co3NiB2 各构型的HOMO轨道与水分子LUMO轨道的能级及能级差Tab. 2 The ΔE between the HOMO orbital of configurations of Co3NiB2 and the LUMO orbital of H2O kJ·mol -1

图4 4 种优化构型的(Co3NiB2)-H结构Fig. 4 The(Co3NiB2)-H structures of 4 optimized configurations
2.3 吸附氢原子后团簇Co3NiB2 析出氢气的解
吸反应前面主要依据前线轨道理论对团簇Co3NiB2吸附氢原子(即催化水析氢反应机制第一步)的难易程度进行了分析.原始团簇模型Co3NiB2在完成第一步反应后将变成(Co3NiB2)-H结构模型(下文中均用M-H代替),各优化构型吸附氢原子后的M-H空间结构如图4 所示.不难发现,吸附上的氢原子均位于团簇Co3NiB2原始优化构型的Co原子上,这说明Co原子是团簇Co3NiB2的潜在催化活性位点,这与该体系前期的研究成果相符[19].诚然,对氢原子吸附过程的研究是十分重要的,但若想完成整个催化析氢反应,氢气的析出过程也同样重要.由析氢反应机制可知,氢气的析出可以分为电化学解吸与化学重组2 种方法.下面便针对M -H 模型的2种方法分别进行解吸过程的难易程度分析.
2.3.1 M-H通过电化学解吸法析出氢气 吸附氢原子后的M-H模型在进行电化学解吸时,电子同样由M-H的HOMO轨道向水分子的LUMO 轨道转移从而使水分子中的氢原子被吸附在M-H模型上,新吸附的氢原子与原有的氢原子结合并以氢气的形式脱离M -H 模型,从而实现M-H模型的解吸过程.当氢气析出后,M -H 模型也将变为原本的Co3NiB2结构,这也就意味着团簇Co3NiB2与水分子进行催化析氢反应时的催化作用发挥完毕.不难发现,采用电化学解吸法析出氢气的反应机制与第一步相似,只不过该解吸过程是将反应底物M换成了M-H.因此,同样可以通过比较M-H各结构模型的HOMO 轨道(EHOMO)与水分子LUMO 轨道(ELUMO)的能级差大小(ΔE)来衡量和预测该过程进行的难易程度.表3 则计算出了M -H各结构模型HOMO轨道与水分子LUMO轨道之间的能级差值.
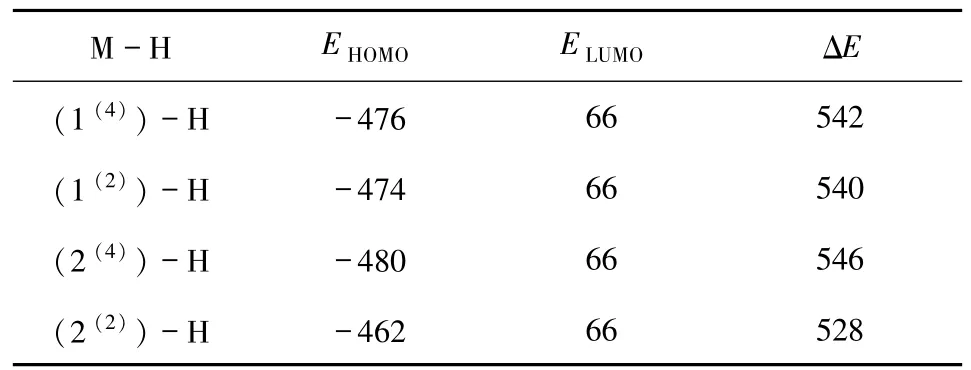
表3 (Co3NiB2)-H的HOMO轨道与水分子LUMO轨道的能级及能级差Tab. 3 The ΔE between the HOMO orbital of(Co3NiB2)-H and the LUMO orbit of H2O kJ·mol -1
综合分析表2、表3 可以发现,当团簇原始优化构型M吸附上一个氢原子后形成M-H结构时,相较于吸附前,其ΔE值明显增大,这说明M-H与水反应的活性有所减弱,其中构型2(4)的增幅最大(Δmax=283 kJ·mol-1),构型1(2)的增幅最小(Δmin=11 kJ·mol-1).比较表2 与表3 中ΔE可知,在第一步反应中EHOMO值最高且最容易给出电子的构型2(4)在解吸过程中的反应活性相较于其他优化构型而言最弱(ΔEmax=546 kJ·mol-1);而在第一步反应中反应活性处于中等水平的构型2(2)在吸附氢原子形成(2(2))-H模型后,其EHOMO值最大且ΔE最小(ΔEmin=528 kJ·mol-1),因而构型2(2)在解吸过程中的反应活性最强.值得说明的是,团簇Co3NiB2各优化构型在吸附氢原子后形成的M-H模型全部满足ΔE<579 kJ·mol-1,说明在实际反应中各优化构型完成解吸反应的活性均较好且相似,这进一步证实了团簇Co3NiB2具有良好的催化析氢活性,其中构型2(2)在解吸过程中反应活性最强.
2.3.2 M-H通过化学重组法析出氢气 吸附氢原子后的M-H 模型通过化学重组法析出氢气时,作为中间体的2 个M -H 分子中的氢原子相互结合,从而以氢气的形式脱离M -H 模型完成解吸过程.同样的,析出氢气后的M-H模型转变为原有的Co3NiB2结构,也就意味着团簇Co3NiB2完成了催化过程,其催化作用发挥完毕.因此,只有当M -H模型的结合能EBE较小时才有助于解吸过程的完成.为了使氢气能顺利析出,在M -H 模型作为反应中间体的基础上对各构型吸附模型的结合能进行比较分析,以此得到团簇Co3NiB2各优化构型的催化活性大小顺序也是非常重要的一步.由表4 可得,图4中所有M-H模型的结合能由大到小依次为

结合2.2.2 节的研究分析可知,能级差越大的构型越不易通过与水分子反应吸附上氢原子,且二者结合越不紧密,这也就意味着相应M-H吸附模型的结合能会越小;构型2(4)极易与水分子反应吸附上氢原子,但这也导致其结合氢的强度太大(即导致对应M-H模型的结合能较大),从而不利于氢气的最终析出,这就降低了构型2(4)的催化活性,而M-H吸附模型结合能均较小的构型1(2)与2(2)在吸附氢原子后的解吸反应中,其上的氢原子均较容易脱离,故该二者有利于氢气的产生.

表4 团簇Co3NiB2 的4 种优化构型吸附氢原子后的EBE Tab. 4 The EBE of 4 optimized configurations of cluster Co3NiB2 after absorbing the H atom
综上所述,无论各优化构型的M-H模型是通过电化学解吸法析出氢气还是化学重组法析出氢气,均证明了构型2(2)在催化水析氢的解吸过程中能展现出最佳的催化活性.此外,2.2 节的讨论又证实了构型2(2)从水分子中吸附氢原子的能力也非常好.由此可得,构型2(2)是团簇Co3NiB2中催化析氢活性最好的结构模型.
3 结论
选取优化后稳定存在的4 种构型为研究对象,并以团簇Co3NiB2催化水解析氢时的反应机制为依据,在前线轨道理论的基础上分别从吸氢反应与解吸反应(电化学解吸法和化学重组法)来深入探究团簇Co3NiB2在催化水析氢时的反应活性,从而分析比较出团簇Co3NiB2各优化构型在促进水析氢反应时的催化活性大小.研究结果如下:
1)团簇Co3NiB2是通过各优化构型的前线HOMO轨道与水分子LUMO 轨道间的电子转移来完成析氢反应中吸附氢原子的过程,且其优化构型的β-HOMO在该过程中占据着较大的优势;
2)在吸氢反应过程中,氢原子均吸附在团簇原始空间结构的Co 原子上,即Co 原子为团簇Co3NiB2的潜在催化活性位点;
3)对团簇Co3NiB2进行多角度分析后得到,构型2(2)不仅在催化水析氢反应中的吸氢过程中表现出良好的反应活性,在形成吸附模型M -H后的解吸过程中更是展现出了绝佳的催化效果.由此可知,构型2(2)是团簇Co3NiB2在催化水解析氢反应过程中具有最佳催化活性的优化构型.
猜你喜欢
科教新报(2021年11期)2021-05-12 19:50:11
原子与分子物理学报(2021年2期)2021-03-29 07:31:26
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08 00:48:08
数理化解题研究(2017年16期)2017-07-21 09:32:29
北京航空航天大学学报(2017年10期)2017-04-20 08:51:23
科学之谜(2016年9期)2016-10-11 08:59:04
中学物理·高中(2016年8期)2016-08-08 09:21:06
山西大同大学学报(自然科学版)(2016年6期)2016-01-30 08:29:23
航天返回与遥感(2014年4期)2014-07-31 17:47:47
无机化学学报(2014年4期)2014-02-28 17:31:08
