
高效液相色谱法同时测定复方磺胺间甲氧嘧啶预混剂中磺胺间甲氧嘧啶及甲氧苄啶的含量
2020-12-31 08:05陈锡龙周泽晓王庆红谢丽丽焦仁刚
中国兽药杂志 2020年12期
陈锡龙,周泽晓,王庆红,谢丽丽,郭 萍,古 海,焦仁刚,罗 杨
(贵州省兽药饲料检测所,贵阳 550003)
1 材料和方法
1.1 仪器设备 Thermo Scientific U3000高效液相色谱仪(配DAD检测器);色谱柱为Hypersil GOLD C18,(5 μm, 4.6 × 250 mm)色谱柱。Agilent 1260 infinity 高效液相色谱仪(配紫外检测器);BP211D型电子分析天平(德国塞多利斯公司);超声波清洗器(KQ-300DE,昆山市超声仪器厂)。
1.2 对照品 磺胺间甲氧嘧啶对照品来源于中国兽医药品监察所,批号:C0031610,含量99.5%。甲氧苄啶对照品来源于中国兽医药品监察所,批号:H0161210,含量100.0%。
1.3 原料药 磺胺间甲氧嘧啶:吴江博霖实验有限公司提供,含量:97.38%;甲氧苄啶:寿康富康制药有限公司,批准文号:国药准字H37020335,批号:A-10111710094,生产日期:20171031,含量:99.3%。
1.4 试验样品 共收集了5批2个不同生产厂家生产的复方磺胺间甲氧嘧啶预混剂样品,样品信息见表1。

表1 试验样品信息Tab 1 Relative information of five Compound SMM Premix samples
1.5 试剂 甲醇、乙腈为色谱纯;水为高纯水;其他试剂均为分析纯。
1.6 方法
1.6.1 色谱条件 色谱柱:Zorbax SB C18(5 μm 4.6×250 mm);柱温为40 ℃;流动相为0.1%磷酸溶液-乙腈(86 ∶14);流速为1.0 mL/min;检测波长为240 nm[5];进样体积为20 μL。
1.6.2 对照品溶液的制备 分别精密称取磺胺间甲氧嘧啶对照品50.43 mg,甲氧苄啶10.14 mg,置同一50 mL量瓶中,加甲醇适量,超声处理10~20 min,取出,放冷,用甲醇稀释至刻度,混匀,即得磺胺间甲氧嘧啶浓度为1.0036 mg/mL和甲氧苄啶浓度为202.8 μg/mL的磺胺间甲氧嘧啶和甲氧苄啶混合对照品贮备液。精密量取上述混合对照品贮备液5 mL置于50 mL容量瓶中,加水稀释至刻度,即得磺胺间甲氧嘧啶浓度为100.36 μg/mL和甲氧苄啶浓度为20.28 μg/mL的混合对照品溶液。
1.6.3 供试品溶液的制备 取本品0.5 g,精密称定,置50 mL量瓶中,加甲醇适量,超声处理10~20 min,取出,放冷,再加甲醇稀释至刻度。另取上述稀释液5 mL,置于50 mL容量瓶中,加水稀释至刻度,摇匀,即得。
产业演变过程不仅通过产业集聚的组织形式对区域产生环境效应,也通过产业网络、产业共生等组织形式对区域生态环境产生影响,相关领域的学者已开展了卓有成效的研究(王锋等,2018)。基于455篇文献计量分析和70篇代表性文献分析,展望地理学、经济学、规划学、环境学、社会学等学科对产业集聚环境效应研究的拓展领域和方向。
1.6.4 标准曲线的建立 精密量取上述磺胺间甲氧嘧啶和甲氧苄啶混合对照品贮备液各0.5、1、2、5、10 mL,分别置于5个100 mL容量瓶中,补足甲醇量为10 mL,再加水稀释至刻度,得含磺胺间甲氧嘧啶浓度分别为5.02、10.04、20.07、50.18、100.36 μg/mL 和甲氧苄啶浓度为1.01、2.03、4.06、10.14、20.28 μg/mL系列混合对照品溶液。精密吸取各系列混合对照品溶液20 μL,注入液相色谱仪,记录色谱图。分别以磺胺间甲氧嘧啶和甲氧苄啶色谱峰面积(Area)为纵坐标, 其相应质量浓度(Amt, μg/mL)为横坐标,绘制标准曲线,求得各自回归方程和相关系数(r)。
1.6.5 含量测定 精密吸取上述1.6.2项中的磺胺间甲氧嘧啶和甲氧苄啶混合对照品溶液及1.6.3项中的供试品溶液各20 μL,注入液相色谱仪,记录色谱图。按外标法定量计算供试品的含量。
2 结果与分析
2.1 色谱图 分别精密吸取磺胺间甲氧嘧啶和甲氧苄啶混合对照品溶液及供试品溶液各20 μL,注入液相色谱仪,记录色谱图(图1)。结果表明:供试品溶液色谱图中,在与混合对照品溶液色谱图相应的位置上,有相同保留时间的色谱峰。
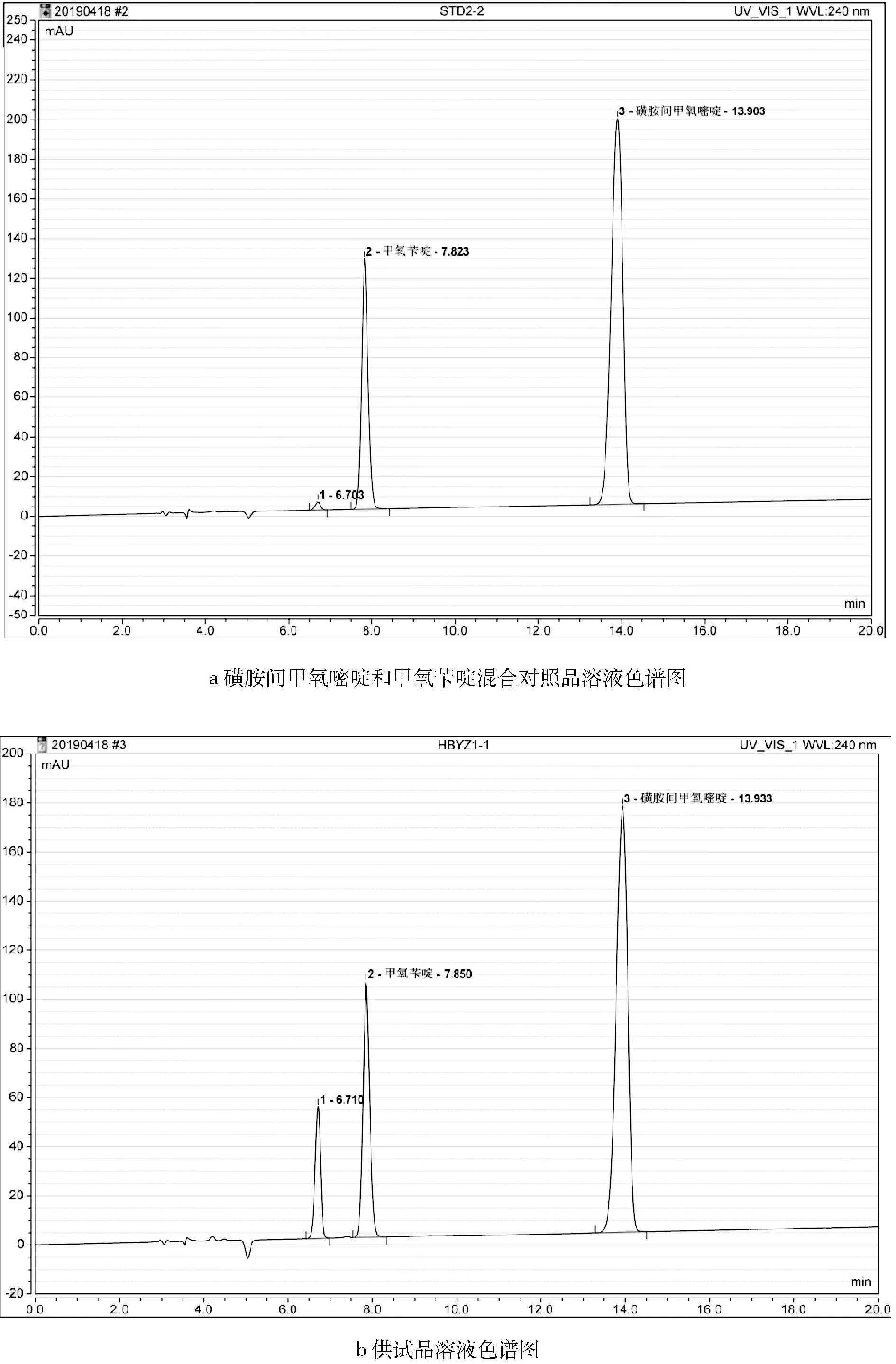
图1 磺胺间甲氧嘧啶和甲氧苄啶混合对照品及预混剂供试品溶液高效液相色谱图Fig 1 HPLC chromatograms of reference substances or Compound SMM Premix sample
2.2 线性范围 分别精密吸取上述1.6.4项中各系列梯度的磺胺间甲氧嘧啶和甲氧苄啶混合对照品溶液20 μL,注入液相色谱仪,记录色谱图。以浓度为横坐标(Amt),色谱峰面积为纵坐标(Area),进行线性回归,得到磺胺间甲氧嘧啶和甲氧苄啶的回归方程。结果表明:磺胺间甲氧嘧啶在5.02~100.36 μg/mL和甲氧苄啶在1.01~20.28 μg/mL内线性关系良好,其线性方程及相关系数见表2。

表2 线性方程及相关系数Tab 2 Linear equation and related coefficient
2.3 重复性 精密量取含磺胺间甲氧嘧啶浓度为100.36 μg/mL和甲氧苄啶浓度为20.28 μg/mL的复方磺胺间甲氧嘧啶混合对照品溶液及含量测定项下编号为SHGY1的供试品溶液各20 μL分别连续进样6次,以测定结果峰面积的相对标准偏差表示试验的重复性,磺胺间甲氧嘧啶色谱峰面积的RSD分别为0.03%和0.13%,甲氧苄啶色谱峰面积的RSD分别为0.10%、0.20%,表明方法的重复性良好。结果见表3。
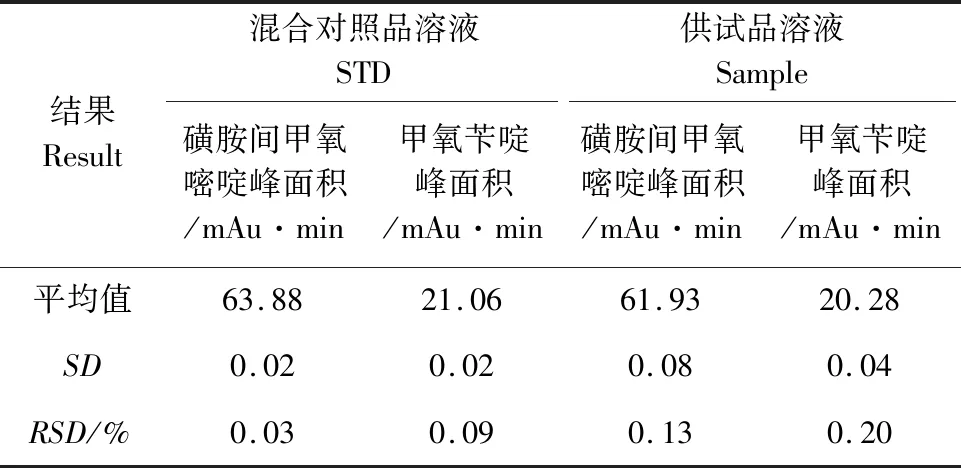
表3 重复性试验结果(n=6)Tab 3 Test data of repeatability
2.4 准确度 以所建立方法的测定结果同原永停滴定法测量结果进行比较,计算5批样品磺胺间甲氧嘧啶和甲氧苄啶两种方法测量结果平均值的相对偏差,结果见表4。结果表明,两种方法测量结果的相对偏差均不大于2.42%,方法具有较好的准确度。
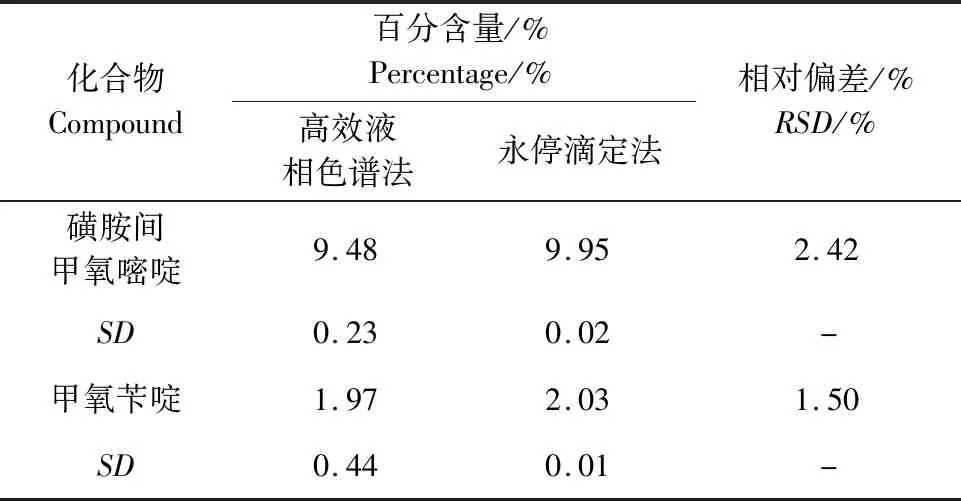
表4 准确度测量结果(n=5)Tab 4 Accuracy results(n=5)
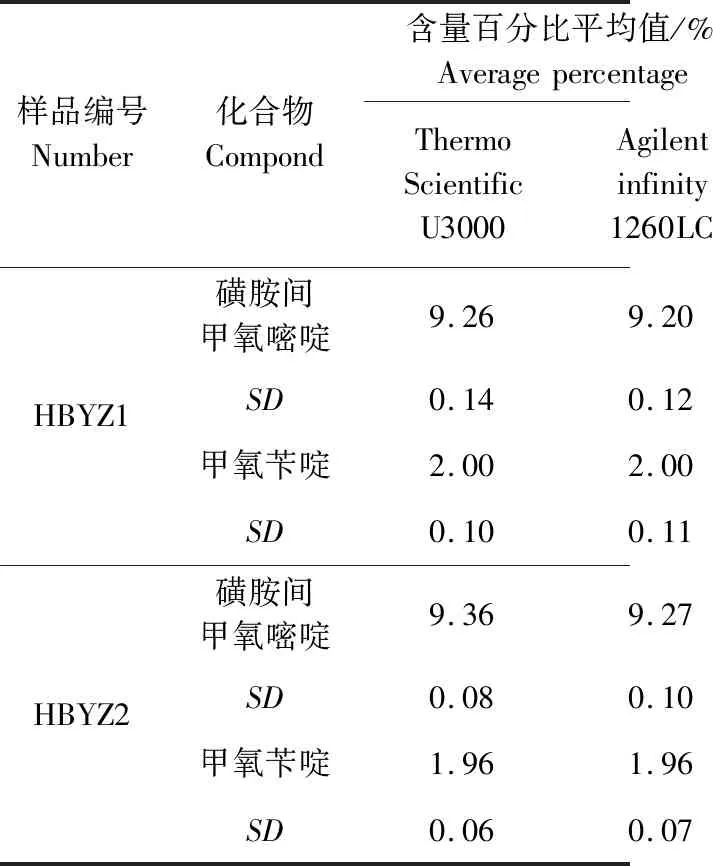
表5 耐用性试验结果(n=3)Tab 5 Durability test results(n=3)
2.6 专属性 在对照品及样品测试色谱图中,甲氧苄啶及磺胺间甲氧嘧啶标准出峰时间附近未发现明显的干扰峰,甲氧苄啶与其前面杂质峰的分离度为4.3,与磺胺间甲氧嘧啶峰的分离度为15.0;同时,经DAD检测器对两个色谱主峰进行识别和定位,均表明方法的专属性良好。
2.7 溶液稳定性试验 精密量取含磺胺间甲氧嘧啶浓度100.36 μg/mL和甲氧苄啶浓度为20.28 μg/mL的复方磺胺间甲氧嘧啶对照品溶液20 μL,分别约于0、24、48 h上机测定,每次进样两针,磺胺间甲氧嘧啶和甲氧苄啶三次测量的色谱峰面积平均值的相对标准偏差为1.36%和1.45%,结果见表6,表明溶液在48 h内稳定。
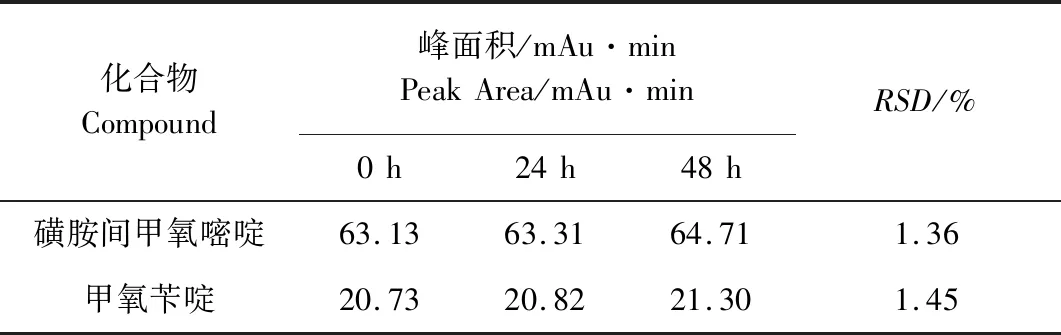
表6 稳定性测量结果Tab 6 Stability results
2.8 添加回收试验 以编号为SHGY1的样品作为本底样品,称取约500 mg,精密称定,精密添加磺胺间甲氧嘧啶原料药(含量为97.38%)约50 mg和甲氧苄啶10 mg(含量为99.3%),使两者的添加量均达到原本底含量的约100%,按照供试品溶液的配制方法制成供试品溶液进行含量测定,共做8个平行。按(实测含量-本底含量)/实际添加量×100%计算添加回收率,测得磺胺间甲氧嘧啶的回收率为98.83%~101.95%,平均回收率为100.53%,RSD为0.91%;甲氧苄啶的回收率为99.02%~103.09%,平均回收率为101.53%,RSD为1.23%,结果见表7。
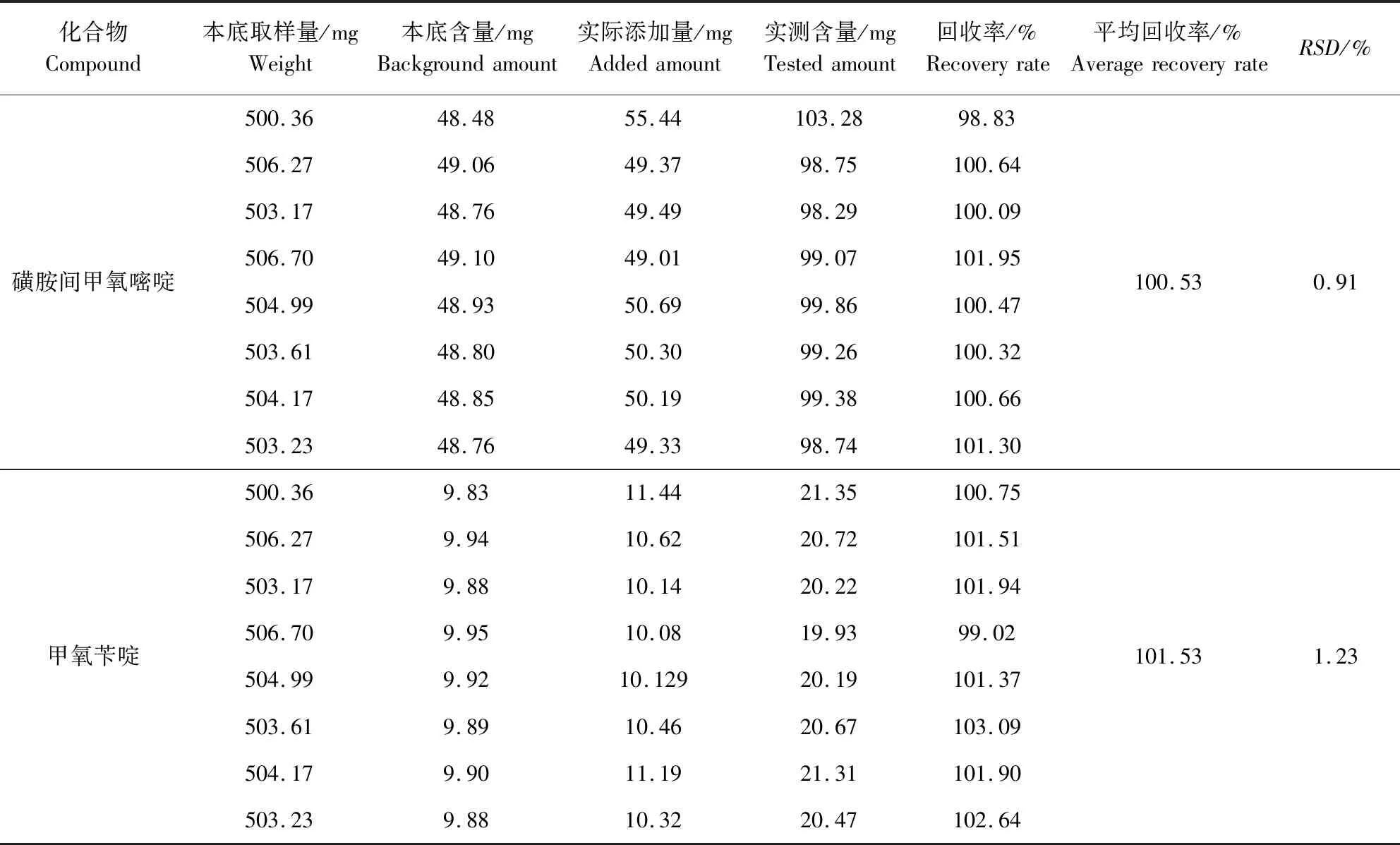
表7 磺胺间甲氧嘧啶、甲氧苄啶回收率测量结果(n=8)Tab 7 Recovery rates of SMM and TMP(n=8)
2.9 样品检测 取5批试验样品,按供试品溶液的制备方法分别制成供试品溶液后进行含量测定,每批样品作三个平行,结果见表8。结果表明,各批次磺胺间甲氧嘧啶的RSD均小于2.0%;甲氧苄啶的RSD相差较大,可能是其含量较低且有部分样品没有混匀的缘故。
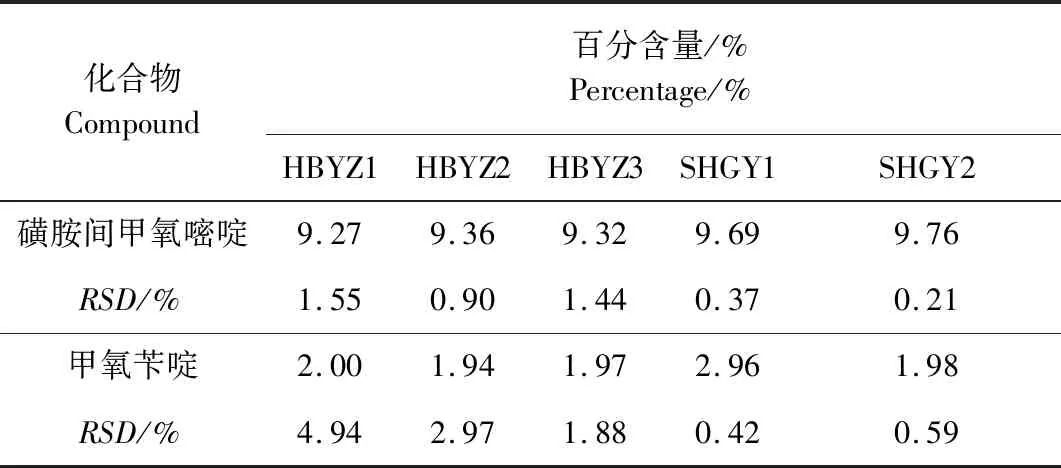
表8 5批样品中磺胺间甲氧嘧啶及甲氧苄啶含量测量结果(n=3)Tab 8 Content determination results of SMM and TMP in 5 samples(n=3)
3 讨论与结论
3.1 色谱条件及检测波长的选择 当前文献报道测量磺胺类药物的高效液相色谱法所使用的流动相系统有0.1%磷酸溶液-乙腈(90∶18,V/V)[6]、乙腈-甲醇-水-乙酸(2+2+9+0.2)[7]、0.017 mol/L磷酸-乙腈(80+20)[8]、磷酸盐溶液-甲醇(65+35)[9]、甲醇-水-三乙胺(200∶799∶1,V/V)(用氢氧化钠试液或冰醋酸调节pH值至5.9)[10-11]等,柱温主要有30、40 ℃,检测波长多为270、240、230 nm。在200~400 nm波长范围内对磺胺间甲氧嘧啶对照品溶液进行波长扫描,发现磺胺间甲氧嘧啶最大吸收峰为295、250、230 nm,在285、270、215 nm处均有较强吸收峰。陈锡龙[12]采用流动相为0.1%磷酸溶液-乙腈(80∶20)检测磺胺间甲氧嘧啶(钠)的含量,认为磺胺间甲氧嘧啶(钠)的出峰时间适当,峰形良好,且0.1%的磷酸溶液也容易配制。由于增加了对甲氧苄啶的检测,流动相的比例也应作出适当调整。检测中发现甲氧苄啶色谱峰的保留时间受流动相比例变化的影响较大,即有机相增加,出峰时间提前,且峰形尖锐;有机相减少,出峰延迟,峰形变钝。研究发现,同时检测磺胺间甲氧嘧啶和甲氧苄啶时,采用流动相0.1%磷酸溶液-乙腈(86∶14)效果最佳。但有专家认为该流动相酸性较强,有可能会损伤色谱柱。实验表明,该流动相的pH值略大于2.0,约为2.05左右,满足一般色谱柱对pH值的要求范围,且自从2013年开始使用0.1%磷酸溶液-乙腈流动相系统研究检测磺胺类药物以来,未曾发生一例色谱柱受到损坏的情况。为使该0.1%磷酸溶液-乙腈流动相系统(以下简称磷酸-乙腈系统)能被更加广泛地应用于检测多种磺胺类药物,研究还考察了其与甲醇-水-三乙胺(200∶799∶1,V/V)(用氢氧化钠试液或冰醋酸调节pH值至5.9)流动相系统(以下简称甲醇-三乙胺系统)检测不同磺胺药物含量的比较情况,发现后者同样能够对多种磺胺有良好的分离,但缺点是洗脱能力差,容易污染色谱柱,从而导致色谱柱柱效快速下降,使用寿命大大缩短。考虑到为保持将来磺胺类药物检测方法的一致性,同时也能够获得较好的分离效果,故选择240 nm作为检测波长,40 ℃作为色谱柱柱温,0.1%磷酸溶液-乙腈(86∶14,V/V)作为流动相。
3.2 样品提取方法的优化 对于样品的提取方法,陈锡龙[13]采用加入1 mL的盐酸溶解处理样品后再加水定容的方法。但盐酸的破坏力极强,可能会对样品造成不可预知的破坏,也存在一定的安全隐患,同时,该方法处理样品后需要迅速进行稀释,否则,磺胺间甲氧嘧啶特别是甲氧苄啶容易析出结晶,导致测量结果出现偏差。用甲醇提取样品或溶解对照品时可获得满意的检测效果,但要注意上机溶液中有机溶剂的比例不宜超过20%,否则,易产生溶剂效应,使甲氧苄啶的峰形变差,甚至出现分叉等,从而影响检测结果。
3.3 高效液相色谱法测量结果比永停滴定法略低的原因分析 使用高效液相色谱法测量磺胺间甲氧嘧啶时,其结果往往要略低于传统的永停滴定法测量的结果。分析认为是磺胺间甲氧嘧啶在贮存过程中会产生一定程度的降解,且其降解产物也属于磺胺类物质,使用永停法进行测量时,仪器无法识别出该产物;而使用高效液相色谱仪进行测量时,该降解产物则能够被很好地识别出来,即在前文图1中磺胺间甲氧嘧啶和甲氧苄啶混合对照品及预混剂供试品溶液色谱图中保留时间约为6.7 min(磺胺间甲氧嘧啶相对保留时间约0.48处)的较小的色谱峰,所以,高效液相色谱法测量结果比永停滴定法略低也就不难理解了。该降解产物经二极管阵列检测器检测,在238.8、265.0、288.6 nm波长处有最大吸收。其相对峰面积甚至可能高达10%以上。但是,关于该降解产物的性质及其对药品质量会产生什么样的影响尚不清楚。
本研究建立的复方磺胺间甲氧嘧啶预混剂含量的高效液相色谱测定法是可行的,该方法简单,准确,精密度好,回收率高,可以作为复方磺胺间甲氧嘧啶预混剂的质量控制标准中同时测定磺胺间甲氧嘧啶和甲氧苄啶含量方法的补充。
猜你喜欢
煤化工(2022年3期)2022-07-08
中国饲料(2022年5期)2022-04-26
合成纤维工业(2021年5期)2021-10-31
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
首都食品与医药(2020年1期)2020-10-21
农药科学与管理(2019年6期)2019-11-23
国外畜牧学·猪与禽(2019年8期)2019-11-11
智富时代(2019年7期)2019-08-16
智富时代(2019年7期)2019-08-16
