
鲁拉西酮的药理学、药动学及临床研究进展*
2020-12-31 08:50:02王莹,詹引,李洁
中国药业 2020年24期
王 莹,詹 引,李 洁
(1. 天津市安定医院,天津 300222; 2. 天津市医学科学技术信息研究所,天津 300070)
第2 代抗精神病药物(SGA)是治疗精神分裂症和情感障碍的有效药物,均具有独特的功效和安全性。鲁拉西酮、依匹哌唑、卡利拉嗪和lumateperone 是美国食品和药物管理局(FDA)批准用于治疗精神分裂症的新型SGA 处方药[1],具有改善阴性和抑郁症状的作用,代谢和心血管等方面的副作用较少,锥体外系反应的风险较低。鲁拉西酮也是FDA 批准用于治疗躁郁症仅有的3 种药物之一[2]。
盐酸鲁拉西酮由日本住友制药公司开发,按用途可以为多巴胺和血清素拮抗剂类[3]。2010 年,FDA 批准用于治疗青少年(13 ~17 岁)和成人精神分裂症;2013 年,FDA 批准了2 个新适应证,单药治疗和作为锂或丙戊酸钠的辅助药物治疗,用于成人Ⅰ型双相情感障碍相关的重度抑郁发作;2018 年,FDA 又批准新适应证,用于治疗10 ~17 岁患者双相Ⅰ型障碍(双相抑郁)相关的重度抑郁症发作。在此综述了近年来鲁拉西酮在药理学、药动学及临床研究方面的进展。
1 药理学
鲁拉西酮是一种苯并异噻唑衍生物,是多巴胺D2、5-HT2A及5-HT7受体阻断剂。在大鼠、豚鼠和人类重组细胞中分析其受体,鲁拉西酮是与多巴胺D2L受体、5-HT2A受体和5-HT7受体具有高亲和力的拮抗剂,与5-HT1A受体具有高亲和力的部分激动剂,与去甲肾上腺素受体有中度亲和力的拮抗剂[4-7]。详见表1。
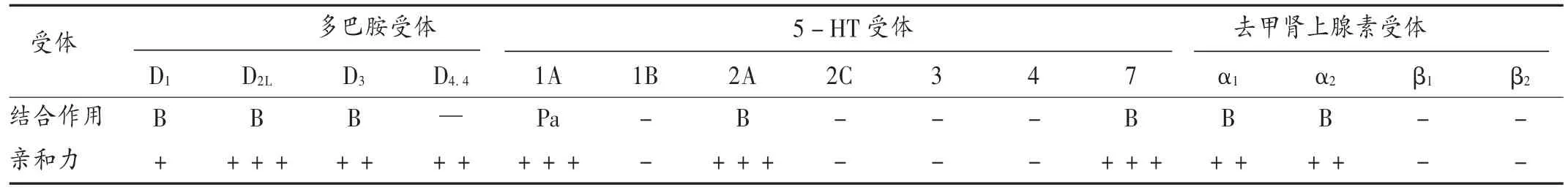
表1 鲁拉西酮与受体的结合作用及亲和力
新一代抗精神病药物的共同特征是D2和5-HT2A受体拮抗作用。5-HT2A受体拮抗作用有助于限制D2受体拮抗剂引起的锥体外系不良反应(如帕金森氏肌肉僵硬和震颤、肌张力障碍、静坐不能)和催乳素升高(与闭经、溢乳、性功能障碍有关),且可能会改善精神分裂症的阴性症状[5]。鲁拉西酮药效学特征的一个相对独特的方面是对 5-HT7受体的有效活性。OKADA 等[8]的研究表明,N-甲基- D-天冬氨酸(NMDA)受体阻断剂MK-801 通过诱导γ-氨基丁酸(GABA)激活丘脑-岛叶谷氨酸能传递,增加额叶皮层中谷氨酸的释放。而鲁拉西酮拮抗丘脑背内侧核(MDTN)中兴奋性5-HT7受体,可抑制MK-801 引起的 L-谷氨酸释放,有助于抗精神病和稳定情绪。鲁拉西酮对以下受体结合亲和力非常低(Ki> 1 000 nmol/L)[4-6]:腺苷 A1和 A2,苯二氮,胆囊收缩素 CCKA和 CCKB,GABAA,α -氨基羟甲基唑丙酸(AMPA),红藻氨酸和谷氨酸NMDA,甘氨酸,组胺H1,毒蕈碱型乙酰胆碱M1和M2,尼古丁,多巴胺和5-羟色胺再摄取位点,这与其在同类药物中更具优势有关。鲁拉西酮不产生由于抗毒蕈碱作用引起的不良反应,如口干、视力模糊、便秘、认知问题;不产生由于抗H1组胺作用引起的不良反应,如过度镇静、食欲增加、体质量增加及代谢综合征和糖尿病;不产生由于显著的5-HT2C拮抗作用引起的不良反应,如食欲增加和体质量增加。
2 药动学
2.1 药动学参数研究
一项临床前研究发现,10 只Beagle 犬口服鲁拉西酮片后,其主要药代动力学参数达峰时间(tmax)、达峰浓度(Cmax)和 0 ~ t h 血药浓度 - 时间曲线下面积(AUC0-t)、0 ~ ∞ 血药浓度 - 时间曲线下面积(AUC0-∞)分别为 (0.8 ±0.5) h、(66.4 ±75.5) ng/mL、(175.9 ±123.7)ng·h/mL、(183.9 ± 124.3)ng·h/mL[9]。
一项针对健康中国人的单中心、随机、平行组、安慰剂对照组和双盲研究评估了单剂量(20,40,80 mg)和多剂量(40 mg/d,连续5 d)口服鲁拉西酮的药代动力学,在20 ~80 mg 剂量范围内单次给药后,血药浓度在1.0 ~ 3.0 h 内达 Cmax,然后双相下降,平均半衰期(t1/2)18.1 ~ 25.5 h,AUC 和 Cmax随剂量按比例增加;连续给药5 d 后达到稳态血药浓度,一个剂量间隔内的 AUC累积指数(AUCτ)为 1.25,小于理论累积系数(1.76),无意外累积,其代谢物(ID-14283,ID-14326 和 ID-11614)也观察到相似结果。该研究认为,中国健康受试者单次和多次口服鲁拉西酮后的药代动力学特征与健康白色人种相似[10]。
2.2 吸收
健康志愿者口服鲁拉西酮后吸收差,而食物可促进鲁拉西酮的吸收。关于食物影响的最确切数据来自两项开放性、随机、交叉研究,研究对象为18 ~65 岁,临床稳定的精神分裂症或分裂情感性障碍患者,体质量指数为19.5 ~ 37.0 kg/m2,且未服用已知是 CYP3A4 抑制剂或诱导剂的药物。这些患者每天单独或与食物同时服用120 mg 鲁拉西酮,第5 天检查 Cmax和给药间隔期间血药浓度 -时间曲线下面积(AUC0-tau)。第 1 项研究(n=16)评估了禁食和3 种餐食(100 kcal/中等脂肪,200 kcal/中等脂肪和800 ~1 000 kcal/高脂肪)对鲁拉西酮生物利用度的影响,禁食状态和800 ~1 000 kcal 餐后 Cmax的几何平均数分别为56.7ng/mL 和123.0ng/mL,平均 AUC0-tau分别为 360.0ng·h/mL 和 752.4ng·h/mL(P < 0.01)。相较于禁食状态,800 ~1 000 kcal 餐后Cmax的几何平均数和平均 AUC0-tau均升高2 倍。进食100,200 kcal 餐后鲁拉西酮的生物利用度显著低于进食 800 ~1 000 kcal 餐后。第 2 项研究(n=26)评估了禁食和 5 种餐食的影响(350 kcal /高脂,500 kcal /低脂,500 kcal/高脂,800 ~1 000 kcal /低脂和 800 ~1 000 kcal/高脂),禁食状态和350 kcal/高脂餐,500 kcal /高脂餐,800 ~1 000 kcal/高脂餐后 Cmax的几何平均数分别为 52.9,161,135,131 ng/mL,平均 AUC0-tau分别为390,743,727,769 ng·h/mL。相较于禁食状态,350 kcal餐后 Cmax的几何平均数升高3 倍,平均 AUC0-tau增加近 2 倍。进食 350,500,800 ~ 1 000 kcal 餐后鲁拉西酮的生物利用度无显著差异,进食低脂和高脂餐后鲁拉西酮的生物利用度无差异。因此,该研究建议口服鲁拉西酮时至少同时进食 350 kcal 餐食,与脂肪含量无关[11]。
2.3 分布
鲁拉西酮主要与血浆白蛋白和α1酸性糖蛋白结合,结合率达99.8%,且与浓度无关。血浆白蛋白随着年龄的增长而减少,α1酸性糖蛋白随着年龄的增长而增加,人群药代动力学分析表明,老年人使用鲁拉西酮不会引起重大的安全性或疗效问题。
一项雄性Sprague-Dawley 大鼠实验研究发现,静脉注射鲁拉西酮 0.5 ~2.5 mg/kg 后,其广泛分布于脑、肝、肾、心脏、脾脏、肺、小肠、肌肉和脂肪组织中,组织与血浆的浓度比范围从 1.06(大脑)到 9.16(脂肪组织)[12]。
2.4 代谢
鲁拉西酮在肝脏被CYP3A4 酶代谢,代谢途径有N-脱烷基氧化、降莰烷环或环己烷环的羟基化、异噻唑基环的 S-氧化、异噻唑环的还原性裂解随后 S-甲基化2 种或多种组合,最常见的代谢途径为 N-脱烷基氧化、降莰烷环的羟基化和 S-氧化。鲁拉西酮被代谢分解为 3 个活性代谢物(ID-14283、ID-14326 和ID-11614),分别占鲁拉西酮的 25% 、3% 和 < 1%[13];2 个无生物活性代谢产物(ID-20219 和 ID-20220)。其中,ID-14283 药理学性质与鲁拉西酮相似,且可能有助于原药的功效,但 t1/2较短(7 h)[14]。
2.5 清除
单剂量口服40 mg 鲁拉西酮,清除 t1/2为18 h。在稳定状态下反复口服给药,清除 t1/2可达到37 h。该药主要通过代谢清除,粪便和尿液中放射性标记代谢物的回收率分别为80%和9%;只有不到0.1%的药物以原形从尿液中清除,可以忽略不计。
一项在雄性Sprague-Dawley 大鼠开展的临床前研究发现,静脉注射鲁拉西酮 0.5 ~ 2.5 mg/kg 后,全身清除率、稳态分布量和 t1/2均与注射剂量无关,分别为22.1 ~ 27.0 mL/ (min · kg),2 380 ~ 2 850 mL/kg 和3.8 ~ 4.4 h[12]。
3 临床研究
3.1 精神分裂症
多项短期、随机、双盲、安慰剂对照临床试验证实了鲁拉西酮(40 ~160 mg/d)治疗精神分裂症的短期疗效显著高于安慰剂,且与剂量无关[15]。一项为期6 周的多中心、随机、双盲、非劣效性研究发现,鲁拉西酮治疗成人精神分裂症的疗效不逊于利培酮,且其对体质量、代谢参数或催乳素水平的影响更小[16]。一项为期22 个月的开放性研究显示,使用鲁拉西酮治疗精神分裂症对患者体质量、葡萄糖、脂质和催乳素的影响最小,且患者的阳性与阴性症状量表(PANSS)总评分均持续改善[17]。一项最新的网状Meta 分析选取了13 项非典型抗精神病药物治疗青少年精神分裂症的随机对照试验,将鲁拉西酮与阿立哌唑、阿塞那平、氯氮平、奥氮平、帕潘立酮缓释(ER)、喹硫平、利培酮和齐拉西酮进行了比较。在PANSS 和临床总体印象量表-严重程度项目(CGI-S)评分上,鲁拉西酮的疗效明显优于安慰剂;与其他口服非典型抗精神病药物相比,鲁拉西酮具有相似的疗效,但体质量减轻及全因停药的风险较低[18]。
3.2 其他精神障碍
一项多中心、随机、双盲、灵活剂量调定、安慰剂对照、平行分组临床试验研究证实了鲁拉西酮单药治疗成人双相Ⅰ型抑郁症的临床疗效和安全性。纳入患者随机分为 3 组,20 ~60 mg/d 鲁拉西酮治疗组(n=166),80 ~120 mg/d 鲁拉西酮治疗组(n=169),安慰剂对照组(n=165),治疗时间均为 6 周。接受鲁拉西酮 20 ~60 mg/d的患者,在1 ~7 d 以20 mg/d 的剂量治疗。接受鲁拉西酮80 ~120 mg /d 的患者接受以下固定鲁拉西酮滴定:1 ~ 2 d,20 mg /d;3 ~ 4 d,40 mg /d;5 ~ 6 d,60 mg /d;7 d,80 mg/d。不同剂量组患者在7 d 后允许在指定的剂量范围内调整鲁拉西酮的剂量,以优化疗效和耐受性[19]。一项随机双盲对照临床试验研究证实,鲁拉西酮辅助锂或丙戊酸钠治疗成人双相Ⅰ型抑郁症的临床疗效和安全性,纳入患者在锂或丙戊酸盐治疗的基础上分别给予鲁拉西酮 20 ~120 mg/d(n=183)和安慰剂(n=165)治疗6 周[20]。以上2 项研究均采用蒙哥马利抑郁量表(MADRS)、临床总体印象量表-双相障碍版(CGI-BP)抑郁严重程度评分对患者进行评估。研究结果显示,与安慰剂组相比,不同剂量鲁拉西酮单独治疗或辅助治疗6 周后均显著降低平均MADRS 总评分和CGI-BP 抑郁严重程度评分,显著改善患者的焦虑症状,生活质量和功能障碍。纳入7 项随机对照研究的Meta 分析表明,与安慰剂相比,鲁拉西酮辅助治疗双相Ⅰ型抑郁症可显著改善抑郁症状,对内分泌和心血管系统的不良反应极小且耐受性较好[21]。
3.3 临床应用的安全性
一项中国健康受试者口服鲁拉西酮安全性和耐受性的研究发现,单次或多次口服鲁拉西酮均未观察到严重不良事件(SAE),也无受试者因SAE 退出研究。常见的不良事件是嗜睡、血液催乳素增加和躁动不安(仅见于多次口服患者),且各项症状都被评为轻度,发生率随剂量增加而升高。不良事件的类型和严重程度与白色人种无异[10]。
以上临床试验中,鲁拉西酮因SAE 引起的停药率与安慰剂相似。鲁拉西酮常见不良反应包括恶心、嗜睡、震颤、静坐不能。与其他SGA 药物(尤其是氯氮平,奥氮平和喹硫平)相比,鲁拉西酮的代谢安全性特别值得注意[15 -16,18 -20]。
4 展望
鲁拉西酮已被证实可有效治疗精神分裂症和双向Ⅰ型抑郁症,不良反应发生率低,安全性、耐受性良好,特别是对体质量、心血管系统、代谢参数或催乳素水平的影响较小,其主要活性代谢产物的药理性质与原药相似且可能有助于原药的功效。但其吸收差,需要指导患者在进餐时同服每日剂量的药物,而使用这种药物的人可能患有严重和持续的精神疾病,坚持饮食指南对其可能具有挑战性。可寻找一种新型的水溶性制剂,如含烟酰胺、苯甲酸钠和柠檬酸钠的速溶片剂,大大提高在水中的溶解度[22];或通过不同的给药途径,如研制鲁拉西酮脑靶向脂质体注射剂[23]、鼻内纳米结构脂质载体[24],将药物直接递送至大脑,这也为鲁拉西酮新剂型的研发提供了方向。
猜你喜欢
临床儿科杂志(2023年8期)2023-09-29 09:38:52
山东冶金(2022年2期)2022-08-08 01:50:42
昆明医科大学学报(2021年10期)2021-12-02 03:24:38
特别健康·下半月(2019年6期)2019-08-01 01:45:35
祝您健康(2019年3期)2019-03-22 08:57:08
时代英语·高一(2018年5期)2018-11-19 10:55:06
作文周刊·小学四年级版(2018年40期)2018-04-09 08:20:04
时代英语·高一(2017年5期)2017-11-14 15:52:20
上海金属(2016年1期)2016-11-23 05:17:28
焊接(2016年8期)2016-02-27 13:05:13
