
SRY阴性46,XX男性性反转综合征1例报道
2020-12-31 05:59洪志丹周春张元珍刘松梅彭建红刘环宇
中国生育健康杂志 2020年1期
洪志丹 周春 张元珍 刘松梅 彭建红 刘环宇
46,XX男性性反转综合征是一种非常罕见的疾病,我国的发病率约1/20 000~25 000[1],但其发病机制不清楚。本文对1例SRY阴性的46,XX男性性反转综合征患者进行系列检查,包含精液分析、血清生殖激素测定、染色体核型分析、SRY基因及Y染色体微缺失检测、外周血全外显子测序,分析其遗传学变化与临床表现的相关性。本研究获得医院伦理委员会批准,且患者知情同意。
临床资料
患者,社会性别男性,28岁,已婚,与现配偶同居6年未避孕未育,于2016年3月就诊。体检:身高177 cm,体重85 kg,男性外观;阴茎长度6 cm(疲软状态),阴囊外观正常,可触及双侧睾丸,体积均约2 ml,质地中,有触痛。生殖系统超声检查提示双侧睾丸发育不良。因外生殖器异常于幼时行尿道下裂成形术、隐睾下降移位术及剖腹探查术,术中未见子宫及附件等女性内生殖器组织,术后多次复查无异常发现。父母体健,非近亲婚配,系独生子女,家族中无类似病史。
常规精液分析结果:精液量1.6~2.2 ml。多次精液常规镜检均未见精子。血清生殖激素检测结果:FSH 33.78 mIU/ml(正常范围:1.27-19.26 mIU/ml),LH 23.51 mIU/ml(正常范围:1.24-8.26 mIU/ml);E229 pg/ml(正常范围:20-47 pg/ml),T 1.66 ng/ml(正常范围:0.75-7.8 ng/ml),PRL 11.09 ng/ml(正常范围:2.64-13.13 ng/ml)。患者FSH、LH均升高,提示高促性腺激素性腺功能不全。染色体核型及Y染色体微缺失分析结果:通过染色体G显带法及染色体非整倍体基因拷贝数变异(copy number variations,CNVs)两种方法所测染色体核型均为46,XX,CNVs检查未发现其他异常。PCR-探针法检测SRY基因及Y染色体微缺失,结果显示Y染色体AZFa(sY84、sY86)、AZFb(sY127、sY134)、AZFc(sY254、sY255)6个位点均缺失,SRY基因阴性。
全外显子测序结果:患者目标区域平均测序深度为97目,目标区域覆盖度达到94.55%,其中测序深度达到20中以上的区域占芯片捕获区域的92.86%,芯片对目标区域覆盖良好。外周血全外显子变异全局总览见图1。数据分析发现在9号染色体上DMRT1(chr9:841647-969090)基因发生了突变,具体为碱基T突变为A (chr9:841971),导致其所编码的氨基酸由S(丝氨酸)变为了T(苏氨酸)。WNT4基因的剪切区发生了突变(chr1:22453975),碱基C突变为G。
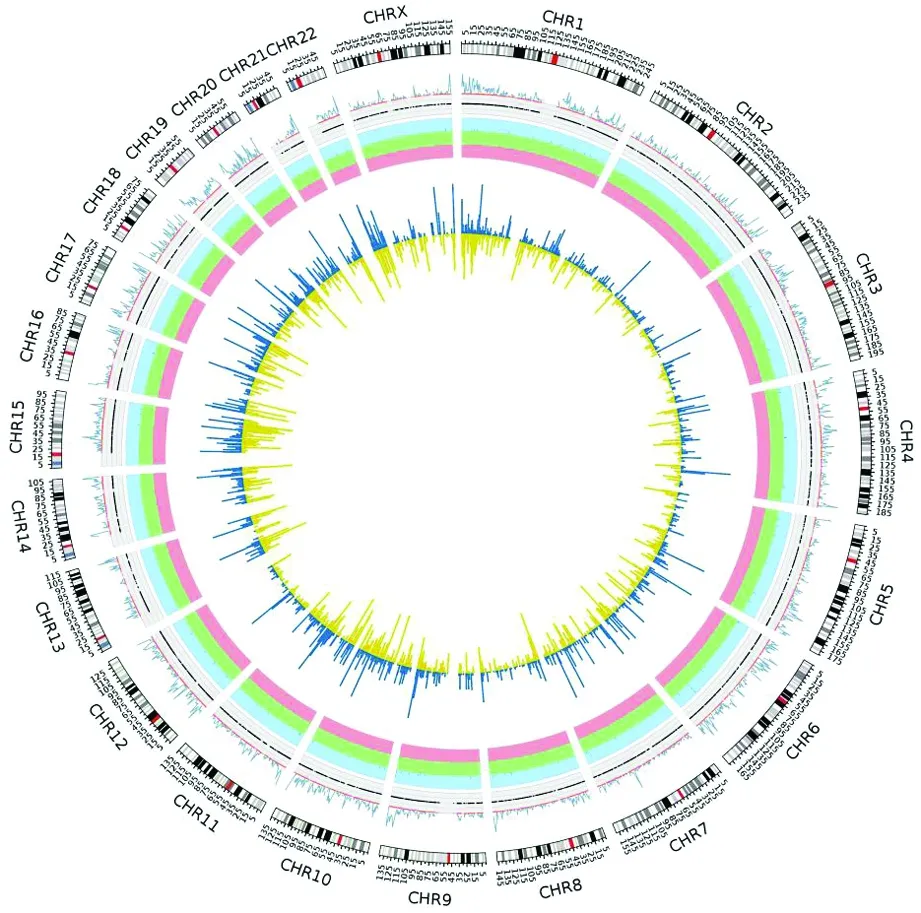
图1 变异全局
从最外层到最内层依次是:(1)第一圈为染色体名称。(2)第二圈为SNP密度(1Mb,曲线),并对密度进行了归一化。(3)第三圈为测序深度,并进行归一化,小于0.25绿色,大于0.75红色,其他灰色。(4)第四圈为不同类型的SV,深绿色代表插入,深蓝色代表缺失,深红色代表扩增(浅绿,浅蓝,浅红为对应的背景色);(5)第五圈为InDel:蓝色为插入,黄色为缺失。
讨论
46,XX男性性反转综合征由1964年由Delachapella[2]首次报道,发病率极低,约90%的患者Y染色体性别决定基因(sex-determining region on the Y chromosome,SRY)为阳性,仅10%左右为SRY阴性[3],其关键特征为“性腺性别与染色体性别不相符”,但目前对此病的了解十分有限。儿童患者常因外生殖器发育异常如尿道下裂、隐睾和生殖器畸形等就诊,而成年男性患者多因不育、性腺功能减退、乳房发育等就诊[4.5]。本病例出生时曾因尿道下裂行手术治疗,当时未作染色体等检查,婚后因不育行精液检查发现无精子症,进而行染色体核型分析从而确诊。因此,对于外生殖器发育异常者进行染色体分析在本病的诊断中具有重要的临床意义。
人类性别决定的关键因素尚未完全阐明,大多数学者认为Y染色体及其中的睾丸决定因子在人类性别决定中扮演关键的角色,但其发病机制并不完全清楚。研究表明,Y染色体易位或是Y染色体缺失伴SRY基因易位是导致本病的最主要原因[6]。但在本研究中,G显带核型分析及CNVs分析并未发现与Y染色体可能相关的标记染色体及DNA序列,且SRY基因、AZFa、b、c均缺失,基本可排除因Y染色体易位或SRY基因易位的致病原因。早期研究中,Mcelreavey K等推测[7],由于促使睾丸发育的基因调控机制发生了隐性突变,致使在无SRY基因存在时睾丸也能发育,此类患者可能为常染色体隐性突变所致,或是常染色体、X染色体上存在未知的性分化决定基因。亦有可能其SRY基因局限于睾丸组织内,而外周血DNA分析检测不出致性反转的基因变异[8],该假设有待于进一步证实。
随着细胞分子生物学研究技术的不断进步,调控人类性别发育的基因逐步被发现,目前已知的有SOX基因家族、DMRT基因、WNT4及RSP1基因等[9]。
性反转相关的报道中较多的为SOX基因家族异常,而对DMRT及WNT4基因异常的报道罕见。DMRT基因簇定位于9p24.3,包括DMRT1、DMRT2、DMRT3基因[10]。DMRT1基因位于9(chr9:841647-969090),它具有一个保守的DNA结合motif(具有特殊功能的蛋白质分子超二级结构),对性腺的发育和配子的形成扮演了决定性的作用。同时,DMRT1编码了一个男性独有的转录因子,在所有脊椎动物中起到调控睾丸分化的作用,该基因突变,可导致生殖细胞无法进行有丝分裂和生殖细胞的分化,影响精原细胞的产生,无法产生精子[9]。WNT4基因为WNT基因家族的一员,位于1p36.12,是一类前卵巢基因/抗睾丸基因,在性腺分化的过程中,其过量表达会抑制睾丸的分化,促进卵巢的分化[11.12]。本病例因不育就诊,检查时发现无精子症,进一步检查发现高促性腺激素性腺功能不全、染色体核型与社会性别不符,不同于常见的生精功能障碍。
本病例高通量全外显子测序结果显示,SOX5、SOX8和SOX10基因的外显子区域发生了单核苷酸多态性(single nucleotide polymorphisms,SNP)突变,但是属于同义突变,其编码的氨基酸序列并未发生改变。而DMRT1基因外显子区域发生了SNP的非同义突变,致使其转录的氨基酸发生了变化,此结果证实了DMRT1基因突变导致无精子症。在传统的观念中,认为无精子症是由于Y染色体的缺失致AZF缺失,导致生精障碍/无精子症,此研究证实AZF缺失并非唯一导致无精子症的原因。本病例发现WNT4基因剪切区的SNP突变,是导致了该基因失活、基因过量表达还是基因表达量降低,通过现有的方法无法明确。但结合患者的临床表现、实验室检查结果以及分子生物学检测结果,我们推测,该基因剪切区的突变,导致患者WNT4基因表达量降低或是WNT4基因失活的可能性较大,因此患者性腺分化为睾丸组织。
在检测结果中,没有发现其他性别分化相关基因的重要有义突变,也未发现SRY基因片段的存在。基于现有的研究结果可以明确,该患者在没有Y染色体及性别决定相关基因的前提下,因WNT4剪切区的SNP突变可能导致其编码产物表达水平的降低或引起WNT4基因失活,导致性腺分化为睾丸组织。同时,其无精子症的原因是由于AZF缺失、DMRT1外显子区域的非同义突变双重作用导致其精原细胞无法分化产生精子。
对于临床诊断的性反转病例,进行基因诊断来明确致病原因有重要的意义。本研究采用高通量全外显子测序技术对46,XX男性性反转综合征进行基因诊断及发现DMRT1基因突变均为国内首次报道。
综上所述,SRY阴性的46,XX男性性反转综合征是罕见的导致男性不育的疾病。本病例通过临床、实验室及分子技术检测方法,在成功找到突变基因后推测了其发生性反转的可能机制。随着不育症发病率的增高,对于无精子症患者建议常规行生殖激素测定、染色体核型、Y染色体微缺失检测,必要时作CNVs及全外显子测序,尤其儿童期如发现外生殖器异常应及时行染色体核型分析,避免误诊漏诊。对于该类有生育要求的患者,因其伴发的睾丸原发性生精障碍患者生育仍是一个难题,可采用供精助孕获得后代。但由于现有方法的局限性,无法确定WNT4基因剪切区的突变对基因表达的具体影响,还需进一步的研究。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中国临床医学影像杂志(2022年5期)2022-07-26
云南医药(2021年6期)2022-01-08
临床检验杂志(2021年10期)2021-11-24
南京医科大学学报(自然科学版)(2021年8期)2021-10-19
川北医学院学报(2021年6期)2021-07-13
中国生殖健康(2020年4期)2021-01-18
湖北农业科学(2015年17期)2015-10-09
湖北农业科学(2014年11期)2014-09-10
现代家庭(1999年7期)1999-06-14
