
锕系元素的非寻常氧化态化学Ⅰ.气相、固相和水溶液
2020-12-30 09:48苏一茂黄闻亮
核化学与放射化学 2020年6期
邓 翀,苏一茂,黄闻亮
北京大学 化学与分子工程学院,北京分子科学国家研究中心,稀土材料化学及应用国家重点实验室, 放射化学与辐射化学重点学科实验室,北京 100871
锕系元素是89号元素锕到103号元素铹共15个金属元素的总称。传统上,前七个元素(锕至镅)统称为前锕系元素,而后八个元素(锔至铹)统称为后锕系元素。锕系元素约占目前元素周期表中元素总数的1/8,并与镧系元素合称f区元素。在锕系元素中,钍和铀在地壳中有一定的分布[1];此外,锕和镤在自然界中也有极少的存量。其他锕系元素最早都是通过人工核反应发现的,它们的合成往往涉及连续中子俘获与β衰变,或者轻核(4He、11B、12C、15N、16O等)轰击过程。局限空间内的高能核爆也可以产生超铀元素,锿和镄最早就是在氢弹爆炸后的辐射落尘中被发现的[2]。所有锕系元素的同位素均具有放射性[3]。这使得它们有着丰富的核反应性,尤其是自发或诱导的核裂变性质。这一方面促成了以铀为主的核能利用,但另一方面也使锕系元素的化学研究必须在一定的辐射防护条件下进行。因此,锕系元素的化学研究,相比于拥有稳定同位素的元素,更为受限。232Th和238U在锕系核素中有着最高的稳定性,其α衰变的半衰期大于109a;同时,它们也是天然钍和铀中占比最高的同位素,因此二者被广泛地用于化学研究[3]。
当前,常规价态的锕系元素配位化学已经不足以满足锕系化学发展的需求。对于非寻常价态的锕系化学的追求不仅可以满足化学家打破边界的好奇心,同时还有着广泛的应用前景。比如,实验上发现将Bk3+氧化至Bk4+有利于锫的分离[4]。本综述(第一部分)将从气相、固相和水溶液等方面勾勒锕系元素非寻常氧化态化学的发展历史与最新进展,首先分析锕系元素电子构型的特点,并给出文中所涉“非寻常氧化态”的定义与范围。对于研究较多的元素,如钍、铀等,将单独进行介绍,而其他锕系元素则分为前锕系与后锕系两类分别介绍。
1 锕系元素的电子构型与氧化态
锕系金属原子及其三价和四价阳离子的基态价电子构型列入表1。对于锕、钍原子,由于6d轨道能量低于5f轨道,电子优先填充在6d轨道上。从镤开始,6d轨道上只占据一个电子,5f轨道开始填占电子。从钚开始,除了5f76d17s2构型的锔和5f146d17s2构型的铹以外,6d轨道不再填占电子,新增的价电子均填入5f轨道。锕系金属原子对应的三价和四价阳离子则具有[Rn]5fn基态电子构型[3]。不同于稀土金属收缩于离子核内部的4f轨道,锕系金属的5f轨道基本不被6s和6p亚层所屏蔽,而5f轨道比4f轨道在原子核外也更加扩展[5]。2000年以来,对相似体系中镧系/锕系金属配合物的对比研究,也证实了5f轨道较4f轨道能更好地参与形成共价键[6-8]。
对于前锕系元素,7s、7p、6d、5f轨道的能量差异不大,这些几乎简并的轨道都有可能参与到化学反应过程中。此外,锕系金属的5fn7s2和5fn-16d17s2电子构型之间的能量差显著小于镧系金属,使得锕系金属还可能以不同的电子构型参与反应。这些特点使得前锕系元素可以在化合物中表现出丰富的氧化态[9]。对于铀及其之前的前锕系元素,最稳定的氧化态是各自的最高氧化态(即失去全部价电子),分别为:Ac(Ⅲ)、Th(Ⅳ)、Pa(Ⅴ)、U(Ⅵ)。对于镎、钚、镅而言,最稳定的氧化态分别为+5、+4、+3[10]。由于5f电子之间并不能有效地相互屏蔽核电荷,因而随着锕系元素的原子序数增大,5f电子与核结合得更加紧密,其能量急剧降低。这一方面使得基态原子的价电子优先填入5f轨道[9],另一方面也使得5f电子在化学反应中更难失去。因此,镎、钚、镅等无法以+7或更高氧化态稳定存在。5f能量骤降带来的影响更突出地体现在后锕系金属离子的氧化还原性质上。除了少数几个元素可以在相对温和的条件下实现变价(如钔、锘的介稳/稳定二价和锫的介稳四价),后锕系元素的化学行为在大部分情况下均由正三价主导。这一点类似于镧系元素,使得后锕系元素与镧系元素在化学性质上非常相近,给核燃料后处理过程中二者的分离造成了困难[11-12]。
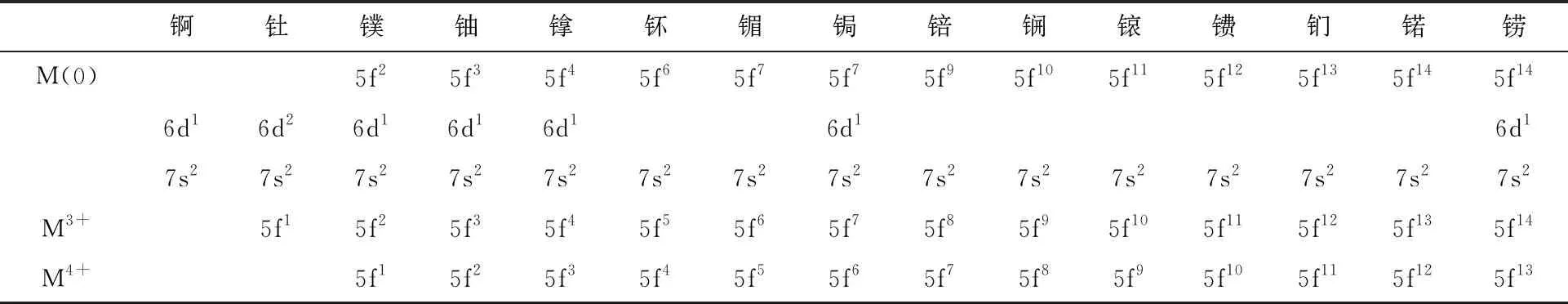
表1 锕系金属原子及三价和四价离子的基态价电子构型Table 1 Ground state electron configuration of M(0), M3+ and M4+ of actinides
三价锕系金属的六配位有效离子半径(r(M3+))[13]及其M3+/2+和M4+/3+电对的标准还原电势(E)[14]列入表2。借鉴于稀土金属非传统氧化态的归类,原则上应当将比二价钐更难还原得到(E(Sm3+/2+)=-1.55 V vs.一般氢电极(NHE))或比四价铈更难氧化得到(E(Ce4+/3+)=+1.7 V vs. NHE)[16]的锕系金属的氧化态作为“非寻常氧化态”。不过由于锕系金属的氧化态变化远比稀土金属复杂,并且其氧化还原电势显著受到环境(溶液酸碱性、配体体系等)影响,因此接下来将展开进行具体讨论。低价方面,E(M3+/2+)从锕到锘递升,其变化范围明显大于镧系金属系列[17]。考虑到绝大部分锕系金属的E(M3+/2+)具有相当大的负值绝对值,将锕系金属的二价认定为“非寻常氧化态”,也包括较易得到的Md(Ⅱ)、No(Ⅱ)以作对比。此外,后锕系元素的E(M4+/3+)均接近或远高于E(Ce4+/3+),因此将从锔开始的后锕系金属的四价及更高氧化态均归为“非寻常氧化态”。考虑到四价钍和镤很难被还原至三价(E(Th4+/3+)=-3.8 V,E(Pa4+/3+)=-2.0 V vs. NHE),将钍、镤的三价也纳入“非寻常氧化态”。
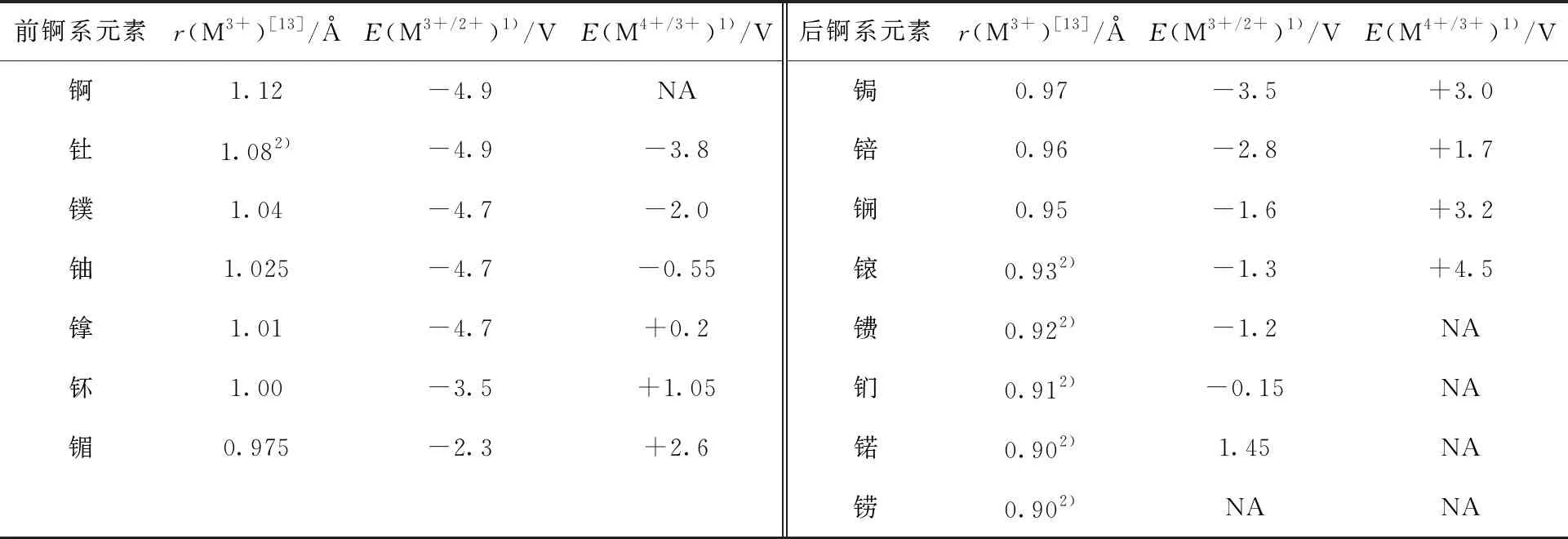
表2 三价锕系金属离子半径与M3+/2+、M4+/3+还原电势(vs. NHE)[14]Table 2 Ionic radii of M3+ and standard reduction potentials for M3+/2+and M4+/3+ (vs. NHE)[14]

综上分析,将Ac(Ⅱ)、Th(Ⅱ)/Th(Ⅲ)、Pa(Ⅱ)/Pa(Ⅲ)、U(Ⅱ)、Np(Ⅱ)、Pu(Ⅱ)、Am(Ⅶ)、M(Ⅱ)/M(Ⅳ)(M=Cm、Bk、Cf、Es、Fm、Md、No、Lr)等视为锕系元素的“非寻常氧化态”,并在本综述中予以讨论。其中包括已经得到结构或光谱等表征确认的氧化态(如Th(Ⅲ)、U(Ⅱ)),也包括当前对其存在尚有争论的氧化态(如Pu(Ⅷ)、Md(Ⅰ))。

表3 锕酰离子相关的M(Ⅴ/Ⅳ)、M(Ⅵ/Ⅴ)、 M(Ⅶ/Ⅵ)还原电势(vs. NHE)1)[14]
2 气相
2.1 钍
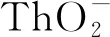
2.2 铀
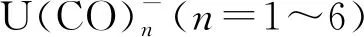
2.3 其他前锕系元素
(1) 镤

(2) 钚
Green等[47]利用溅射设备在低温稀有气体基质中分离了PuO等钚氧化物,并测定了红外光谱。Domanov等[48]利用气相热色谱技术研究了微量钚与氧气在氦载气中的反应,得到的易挥发物种被指认为PuO4。虽然PuO4和PuF8、PuO2F4均被认为是可能的Pu(Ⅷ)目标分子[49],但表征数据的缺失使得PuO4存在的真实性存疑。Zaitsevskii等[50]的计算研究表明,PuO4的稳定性高于AmO4,但不及双金属化合物PuAmO7。利用稀有气体基质的稳定化作用有可能在4~6 K得到PuO4、AmO4[51],而Gong等[52]关于IrO4的气相合成也为此提供了技术参考。关于PuO4的真实电子结构,Huang等[53]指出,尽管钚和钌、锇一样具有八个价电子,但PuO4并不存在与RuⅧO4、OsⅧO4相似的稳定电子构型。通过多种量子化学计算方法,他们发现PuO4的基态是C2v对称的(PuⅤO2)+(O2)-五重态,而D4h对称的PuⅧO4只是亚稳态。类似的“降价”现象同样出现在PuOnF8-2n(n=0~4)体系中,但这一体系中不同电子结构的能量差值更小[54]。
(3) 镅

2.4 后锕系元素
(1) 锔
Smith等[56]最早发现Cm2O3在高温下的气相解离产物主要是CmO。CmO的等电子体,CmF+,也曾在Cm+与六氟丙烯的气相反应中被发现[57]。通过引入硝酸根配位,Kovcs等[58]首次在气相中稳定了五价锔酰离子:[CmO2(NO3)2]-。在气相能与水分子反应,首先形成的[CmO2(H2O)4]+经氧交换过程转化为高价锔氟化物的气相化学十分复杂,虽然早期的热色谱研究发现了CmF6、CmOF3和EsF4存在的证据,但其中锕系金属的氧化态却无法清楚指认[60]。Domanov等[61]利用气相热色谱技术,研究了微量CmHx(2≤x≤3)在氦载气中与O2的反应,得到的易挥发锔化合物因与PuO4有某些相似的性质而被指认为CmⅧO4。但是,由于缺少对金属氧化态的直接表征,这一结论尚存在争议[51]。
(2) 锫、锎、锿与镄、钔、锘、铹

3 固相
3.1 钍
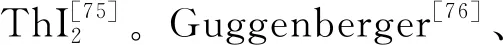
钍的低价氧化物尚无在固相存在的可靠证据,但钍与硫、硒、碲可以形成多种低价化合物[78]。以硫为例,真空中高温加热ThS2可以获得Th7S12;ThS2与钍氢化物在还原气氛下高温加热可得到ThS;在钍氢化物和H2S的反应产物中发现了Th2S3[79]。在加热条件下,钍粉或钍屑还可与氮气或氨气反应生成ThN[80],其薄膜具有典型的金属性[81]。钍单质在高温下可与Th3P4反应生成ThP[82],而ThAs和ThSb则可由单质直接反应得到[78]。
3.2 铀
2018年Havela等[83]利用反应溅射沉积的方法合成了UH2薄膜,并发现其是一种金属性的铁磁体。X射线光电子能谱(XPS)研究表明,UH2和UH3的电子结构有一定的相似性。低价铀的氧化物(如UO)早在1950年就被发现[84],其中UO相需要与UC、UN等共存才能稳定。但是,到目前为止UO在固相的结构尚不明确[85]。Eastman等[79]发现,通过将US2与铀氢化物混合加热至2 000 ℃以上,或利用铀氢化物与H2S在一定条件下反应,均可以制备US。此外,Kulyukhin等[86]还发现,在氯化物熔盐中,Nd2+可以还原U3+。Langridge等[87]向USb中引入Te,发现得到的物质中有UTe相,并通过磁学研究推测有U2+存在。U2+在CaF2等氟化物晶体中的掺杂[88-89],以及U2+与Gd2Cl3共结晶的研究也有相关报道[86]。但是,目前尚没有充分的证据可以指认这些固相化合物中铀的氧化态。
3.3 其他前锕系元素
(1) 锕
尽管1970—1980年代的系列研究揭示了绝大部分f区元素都能以+2氧化态存在于氯化物熔盐中,但二价锕目前尚无在熔盐中形成的证据[90]。
(2) 镤
金属镤在氢气中250 ℃加热可以得到PaH3,而调节反应温度和氢气的压力可得到组成为PaHx(x=1.3~3或2~3)的相或Pa3Hx(x=4~5)的固溶体[91]。PaI3是目前唯一报道的低价镤卤化物,在300 ℃下加热PaI5即可得到。它具有和CeI3相似的粉末X射线衍射(PXRD)图谱[92]。低价镤的氧化物还没有独立报道,但Zachariasen等[93]曾发现金属钡还原PaF4的副产物之一是PaO,并发现其具有NaCl型结构。此外,利用PaI5热解离反应制备的新鲜金属镤,能够与氮气在1 100 ℃反应得到PaN[94];而从镤与砷的单质出发,经过碘的化学气相输运反应可以得到PaAs[95]。
(3) 镎、钚
金属镎能与氢气在不同温度、压力条件下反应。随着氢气含量的增加,产物逐渐由NpH2+x(x=0~0.7)转变为NpH3[96-97]。金属钚与氢气的反应则生成组成连续的固溶体,并不存在PuH2、PuH3的纯相[98]。磁学研究、XPS/俄歇电子能谱(AES)、中子粉末衍射等结果表明,形式上的低价或混价钚氢化物中钚仍以三价形态存在,其准确的电子结构可描述为Pu3+(H-)x(e-)3-x(x<3)[99-101]。Mikheev等[102]曾报道在SrCl2熔盐中用PrCl2还原PuCl3,得到了PuCl2,并推测出Pu3+/Pu2+电对的还原电势为-2.59 V(vs. NHE)。但后来关于Pu(Ⅲ)、Np(Ⅲ)在熔盐中的电化学还原研究均不支持二价中间物种的形成[103-105]。将Np2S3与金属镎混合后高温加热[106],或者进行镎和硫单质的蒸气反应,均可以得到具有NaCl型结构的NpS[107]。此外,PuS则可以通过钙蒸气还原PuF3或钚粉与H2S反应制得[98]。
(4) 镅
在加热条件下,金属镅与氢气反应生成AmH3和AmH2+x两种氢化物,后者与NpH2+x和PuH2+x同构[108]。XPS研究确认了AmH2+x中二价镅的存在[109]。Edelstein等[110]曾将Am2+掺杂到CaF2晶体中,并对其进行了电子顺磁共振(EPR)与可见吸收光谱的表征。Baybarz等[111-112]随后利用金属镅与卤化汞的置换反应,合成了AmCl2、AmBr2与AmI2,并通过磁学测量确认了其中存在Am2+。镅的低价氧化物AmO有可能通过金属镅和Am2O3的归中反应得到,而AmH3与硫单质、硒单质或碲化物共热,则可以得到AmS、AmSe或AmTe[20]。理论计算表明,在这些硫族元素化合物中,仅AmTe中存在Am2+,而且其中镅的半充满5f7电子构型的能量也仅比5f66d1电子构型(其中6d电子离域到导带中)的能量低0.1 eV[113-114]。在熔盐体系中,有较多关于二价镅的研究:比如,金属镅在NaCl-KCl熔盐中与PuCl3反应可以得到AmCl2[115];Am(Ⅲ)在LiCl-KCl、NaCl-2CsCl等熔盐中的电化学还原反应会经历Am(Ⅱ)的形成[116-117]。Am(Ⅱ)在熔盐中的易得性与稳定性,已经在镅与锔和镧系金属的新型分离技术中得到了利用[118]。
3.4 后锕系元素
(1) 锔
1970年,Bansal等[119]通过在纯氢气中加热金属244Cm,得到了首例锔的氢化物。通过比较其与MH2+x(M=Np、Pu、Am)的XRD图谱,他们认为产物是CmH2+x(c≤x≤0.7,晶胞参数c以nm为单位)。Gibson等[120]后来利用锔的更稳定核素248Cm制备了两种晶胞参数不同的氢化物,248CmH3-δ和248CmH2+x,并通过与镧系金属氢化物体系的对比确认了锔的二氢化物的存在。Damien等[121]还发现,金属锔与硫族元素单质在高温下反应下能形成CmA(A=S、Se、Te)等二元硫族元素化合物,但无法得到纯相,而在较低温度下加热,得到的产物是三价锔的化合物。
四价锔化合物都是以氧化物或氟化物形式存在[122]。1955年,Asprey等[123]利用400 ℃下CmF3、Cm2(C2O4)3在氧气中的燃烧反应制备了244CmO2。Morss等[124]对248CmO2进行了中子衍射研究,结果表明化合物的组成为CmO1.99±0.01,但其有效磁矩(3.36±0.06)μB却符合Cm(Ⅲ)的特征。后来的计算研究表明,在CmO2中,锔的氧化态处于+4(f6)和+3(f7)之间,其f电子相比前锕系金属的二氧化物有着更高的离域特征[125]。
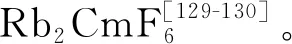
(2) 锫
1972年,Fahey等[133]在石英毛细管中加热金属锫和氢气,得到了组成为BkH2+x(x<1)的锫的氢化物,属立方晶系。Gibson等[134]进一步深入研究了金属锫和氢气的反应体系,发现在600 ℃加热条件下生成BkH2+x,而在300 ℃下则生成六方晶系的BkH3-δ。他们据此确认了Bk(Ⅱ)的氢化物BkH2+x的存在,并认为x取值可能在-0.2~+0.3。
固相中四价锫的稳定性较高,BkO2、BkF4、Cs2BkCl6在1980年代前已得到报道[135]。BkO2具有CaF2型面心立方结构,其价带XPS谱主要体现为5f电子发射的特征[136]。Asprey等[130]利用氟气作氧化剂制备了BkF4。Morss等[137]发现,将Bk(OH)4溶解在浓盐酸后与CsCl反应可以得到Cs2BkCl6,其与Rb2MnF6具有相似结构;而四甲基氯化铵可以替代CsCl作为沉淀剂,得到的[N(CH3)4]2BkCl6具有更低的溶解度和更高的稳定性[138]。此外,尽管在早期的Bk(Ⅳ)共沉淀实验中,经常引入碘酸盐作为沉淀剂[139-140],但BkⅣ(IO3)4的合成直到2017年才由Albrecht-Schmitt等实现。他们发现,向砖红色的Bk(OH)4中加入碘酸,最终可以得到两种晶体,金色块状的Bk(IO3)3与柱状的Bk(IO3)4,其中后者是目前唯一得到单晶结构表征的Bk(Ⅳ)化合物[141]。在这两种碘酸根配合物中,Bk(Ⅲ)和Bk(Ⅳ)分别处在扭曲的三帽三棱柱(九配位)与近乎标准的四方反棱柱(八配位)中心。显微固态光谱仪显示,Bk(IO3)3和Bk(IO3)4在荧光谱和吸收谱上存在显著差异,进一步证实了其中锫具有不同的氧化态。
(3) 锎
目前普遍认为锎能以+2、+3、+4三种氧化态存在于固相中[142]。1968年,Cohen等[143]发现Cf(Ⅱ)可能存在于固相的证据。在加热条件下,用氢气处理含有Cf3+的EuCl3固体,产物溶于水后可与硫酸根离子形成可能为Eu(Cf)SO4的共沉淀。Mailen等[144]随后也在熔盐/液态金属体系中发现了Cf(Ⅱ)存在的证据。Mikheev等[145]则发现,金属镁能在乙醇中还原Cf3+和Sm3+的混合氯化物,而生成的Cf2+能与SmCl2共结晶。后续的研究表明,SrB4O7基质也可用于稳定Cf(Ⅱ)[146]。1972年,Peterson等[147]在加热条件下,用H2还原CfBr3,制备了首例纯的Cf(Ⅱ)化合物CfBr2。在加热条件下,CfBr3能自发分解产生CfBr2,并生成溴单质[148]。CfI2也可通过氢气还原CfI3、或热还原的方式制备[149]。尽管CfCl3的还原要困难得多,但仍有报道:在加热条件下,用氢气还原CfCl3可得到CfCl2,产物得到了固态光谱表征[142]。此外,Gibson等[150]在氢气中加热金属锎,得到CfH2+x,并且发现在常规的合成条件下并不会形成CfH3。研究认为,锎在其氢化物中的真实价态可能介于+2到+3之间[151]。
Haire等[152]用氟气处理CfCl3·xH2O、CfF3或Cf2O3得到了CfF4。三元氟化物MCfF5、M2CfF6、M3CfF7和M7Cf6F31(M为碱金属)被预测能够稳定存在,并且相比CfF4有着更高的热稳定性[142]。Baybarz等[153]利用氧气或铂催化裂解出的原子氧对Cf(Ⅲ)进行氧化,发现在高温下可以生成CfO2。锎的混价氧化物Cf7O12也曾被报道,它在750 ℃以上会逐渐失氧而转化成Cf2O3[142]。
(4) 锿
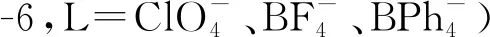
(5) 镄
1972年,Mikheev等[158]发现,用镁还原Fm3+和Sm3+的混合氯化物,得到的Fm2+可与SmCl2共结晶。随后,他们在乙醇溶液中用Yb2+还原Fm3+,使生成的Fm2+与SrCl2共结晶,并通过实验数据估算出E(Fm3+/Fm2+)=E(Yb3+/Yb2+)±0.02 V[159]。基于此,他们设计了一种基于Yb(Ⅱ)-乙醇-水溶液体系的Fm2+与NaCl共结晶的快速分离方法,可以在10 min内将镄从锎、锿与镧系元素等构成的混合物中分离,单次结晶步骤分离因子可达103至104量级[160]。TmI2可以在四氢呋喃(THF)中还原Fm(Ⅲ),产物与[Sr(18-c-6)]I2反应可得到[Fm(18-c-6)]I2[90]。此外,在LiCl-NdCl2-NdCl3熔盐体系中,Fm(Ⅲ)和Es(Ⅲ)也可被还原至+2氧化态[160]。
(6) 钔、锘、铹
二价钔、锘在固相存在的证据来自于共沉淀实验。Hulet等[161]首先在BaSO4共沉淀实验中确认了Md2+的存在。1979年,他们又用示踪量256Md和SmF2/SmF3、SmCl2、RbCl或Rb2PtCl6等进行共沉淀反应,发现钔离子具有和Fm2+、Eu2+、Sr2+相似的性质,而不同于Cs+。据此,他们认为其中钔为Md2+,并否认了该条件下Md(Ⅰ)的存在[162](见4.4节)。锘的稳定二价形态是在LaF3或BaF2共沉淀反应中确定的[163]。对NoH2、LrH2等的理论计算表明,锘、铹的化合物可能分别与镭、铊的化合物具有相似性[164]。
4 水溶液
4.1 钍和铀

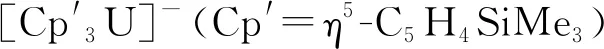
4.2 钚
Tananaev等[173]设计了一系列合成与反应性实验,以确认Pu(Ⅷ)的真实性。比如,他们发现在2 mol/L NaOH溶液中,电解Pu(Ⅵ)也可以得到含有Pu(Ⅷ)的溶液。钚的(超)高价混合溶液可以将Np(Ⅵ)氧化至Np(Ⅶ),而这一氧化过程中既有Pu(Ⅶ)也有Pu(Ⅷ)的参与。不过,他们也指出,不能完全排除化学氧化或电化学氧化形成的“Pu(Ⅷ)”是Pu(Ⅶ)过氧化物的可能性[173]。Tsushima[174]利用DFT计算估计Pu(Ⅷ)/Pu(Ⅶ)的还原电势,发现其值在+1.06 V到+4.36 V范围内变化。其数值取决于所涉及物种的Pu-O键数目,以及Pu的配位数与配位模式,这突显了高价钚溶液体系的复杂性。2014年,Kiselev等[175]发现,在溶液相中,离子型Pu(Ⅷ)物种可能与中性PuO4分子存在平衡。利用CCl4或CHCl3可以萃取水相中的PuO4,但此时七价物种仍然留在水相中。CCl4和CHCl3萃取液的吸收光谱表现出类似OsO4、RuO4等高价金属氧化物的特征,其中332 nm处的最大吸收峰被指认为PuO4的π(O)→Pu荷移跃迁峰。利用XeF2的饱和CCl4或CHCl3溶液与PuO3·xH2O固体接触,也能得到具有相似光谱学特征的Pu(Ⅷ)溶液。
4.3 其他前锕系元素
(1) 锕
1983年,Yamana等[176]研究了水溶液中Ac(Ⅲ)的放射极谱还原化学。通过向体系中引入18-c-6,半波电位有非常大的移动,这被归因于形成了Ac(Ⅱ)-冠醚配离子。通过对离子半径的估算,其中Ac2+的基态价电子构型被推测为6d1。考虑到Ac3+/2+的还原电势估算值为-4.9 V(vs. NHE)[14],Ac(Ⅱ)能否在水溶液中存在尚需要更多表征结果予以确认。
(2) 镤
Guillaumont等[177]通过理论计算发现,6d电子能稳定Pa(Ⅱ)但对Pa(Ⅲ)有去稳定化作用。因此,Pa3+、Pa2+的基态电子构型分别为5f2与5f26d1。对于Pa3+,其5f2构型与5f16d1构型之间能量差较小。因此,Pa3+可以较容易地从5f2基态转化为5f16d1的激发态,随后失去d电子而被氧化成Pa(Ⅳ)。据推算,Pa4+/3+的还原电势为-2.1 V(vs. NHE),而Pa3+/2+的还原电势则为-3.7 V至-4.4 V(vs. NHE)。
(3) 镅
在前锕系元素中,由于Am2+的半充满5f7电子构型,镅的M3+/2+还原电势负值最小(-3.7 V(vs. NHE))[178]。1976年,Pikaev等[179]对含有叔丁醇(氢氧根自由基的清除剂)的Am(ClO4)3水溶液进行脉冲辐解,并通过对产物的吸收光谱的分析得到了Am2+存在的证据。Sullivan[180]、Gordon[181]等同期报道了类似的结果,并展示了更详细的辐解与动力学实验过程。Shilov等[182]认为,辐解形成的Am2+在水溶液中重新被氧化的过程可能与Yb2+、Tm2+等相似,即经历了水合离子的敏化、激基缔合物的形成与分解等步骤。

4.4 后锕系元素
(1) 锔

(2) 锫

近年来,受益于毫克级249Bk生产技术的实现以及表征技术的进步,对溶液中Bk(Ⅳ)行为的研究得以深入[209]。2017年,Deblonde等[210]与合作者发现,在温和条件下,由嗜铁素衍生出的3,4,3-LI-(1,2-HOPO)配体(图1)可以利用分子内的四个氮-羟基吡啶酮(HOPO)单元,实现对金属中心的八齿螯合,从而稳定水溶液中的Bk4+。其中,Bk(Ⅳ)的价态得到了质谱、UV-vis吸收谱表征与DFT计算的支持。液相色谱-质谱(LC-MS)的实验结果表明,这种仿生配体不仅可以实现对Bk(Ⅳ)的高度结合,还能有效地将Bk(Ⅳ)从含有Zr(Ⅳ)、Ce(Ⅳ)、Pu(Ⅳ)、Th(Ⅳ)等其他四价金属离子的混合体系中分离出来[210]。

图1 配体3,4,3-LI-(1,2-HOPO)结构[210]Fig.1 Structure of 3,4,3-LI(1,2-HOPO) ligand[210]
(3) 锎
Cf3+/2+的标准还原电势据估算为-1.6 V(vs. NHE)[14]。1968年,Cohen等[143]利用硫酸盐沉淀实验给出了Cf2+在水溶液中存在的间接证据,他们发现Eu2+能还原Cf3+,但Cr2+不能。Gunningham等率先尝试了水溶液中Cf3+的电化学还原,但由于窗口设置的问题,并没有观察到Cf2+的形成[211]。随后,Friedman等[211]的放射极谱还原研究给出了Cf2+存在的间接证据。1981年,Musikas等[212]发现了水溶液中Cf(Ⅲ)在滴汞电极上的分步还原。不久后,Sullivan等[213]通过Cf(ClO4)3水溶液的脉冲辐解研究给出了Cf2+溶液的时间分辨吸收谱。Friedman等[211]还在乙腈中探究过Cf(ClO4)3的放射极谱还原,发现Cf(Ⅲ)和Sm(Ⅲ)有十分相似的还原性质。2019年,Marsh等[214]在THF中研究了锎-穴醚体系的还原化学,发现穴醚的引入提升了Cf(Ⅱ)的相对稳定性,还观察到[Cf(crypt)]3+(crypt=穴醚[2.2.2])的电化学行为与Sm3+和Yb3+的对应配离子有相似之处。相比于还原,Cf3+的氧化难度更大[215]。Cf4+/3+的还原电势估算值为+3.2 V(vs. NHE)[14],故时至今日仍然鲜有关于Cf(Ⅳ)溶液化学的研究[188]。Payne等[204]曾经报道在乙腈溶剂中三苯基氧胂可以稳定Cf(Ⅳ),但缺少足够的表征结果来证实Cf(Ⅳ)的存在。
(4) 锿、镄、钔、锘、铹
超锎元素的正四价离子尚无在溶液中存在的证据[188],其形成难度从Es4+/Es3+高达+4.5 V(vs. NHE)的还原电势可见一斑[14]。因此,即便是HOPO类配体也很难稳定Es(Ⅳ)[216]。锿、镄、钔的M3+/2+的还原电势估算值分别为-1.3、-1.2、-0.15 V(vs. NHE),而No3+/2+的还原电势骤升至+1.45 V(vs. NHE),这是由于No2+具有全充满的5f14壳层结构[14]。
Es2+、Fm2+的溶液化学研究均与电化学测量相关。比如,在硫酸根离子存在的条件下,水溶液中Es3+的极谱还原研究给出了Es(Ⅲ)→Es(Ⅱ)→Es(0)的分步还原机理[217-218]。钔是第一个被发现能以正二价形态在水溶液中保持稳定的锕系元素。锌粉、锌汞齐以及Cr2+、Eu2+、Yb2+等均能将Md3+还原成Md2+,而V3+/2+电对能在水溶液中与Md3+/2+电对实现氧化还原平衡[161, 219]。流动电解色谱法测得的Md3+/2+还原电势为(-0.16±0.05) V[220]。1976年Mikheev等[221]宣称在乙醇中,用Eu2+或Yb2+可以将Md3+还原成5f14电子构型的Md+,并用CsCl或RbCl实现了Md+的晶格载带。这一发现随后遭到了Samhoun与David等的质疑,他们仔细重复了Md3+的电化学实验,认为在Md2+→Md0的还原过程中并没有Md+的生成[222-223]。对“Md(I)是否存在”这一问题学术界一直存有争论[17, 90, 224-226]。
与Md(I)同样具有5f14电子构型的No2+,则是锘在水溶液中最稳定的离子形态。这一特性使得最早宣称合成102号元素的瑞典斯德哥尔摩大学团队,在利用其发展的基于三价离子分离技术的锕系纯化工艺时,无法获得102号元素(锘)的纯品[227]。Maly等[163]最早发现No2+和碱土金属离子在色谱、共沉淀性质上的相似之处,而用Ce4+氧化之后得到的No3+与其他三价锕系金属离子则有相似性质。No2+的结构化学与配位化学随后得到了研究[228-229],而流动电解柱色谱技术的发展则为调控锘的氧化态提供了可能[230-231]。对于铹而言,由于可供应核素较短的半衰期,仅有几例尝试还原Lr3+的研究被报道。但是,目前在水溶液中还没有发现低价铹离子(如Lr2+)存在的证据[232-234]。
5 总结与展望
近年来,锕系元素的非寻常氧化态化学的研究已经取得了重大突破。举例来说,在1967年以前,人们对锕系元素+2氧化态的了解仅限于CaF2基质中掺杂的Am(Ⅱ),而六价以上的氧化态则尚未涉足[235-236]。到了1980年代后期,人们利用实验结合理论的方法估算了几乎所有锕系元素(除了铹)的M4+/3+和M3+/2+的还原电势值,并对七价镎、钚的合成与化学性质有了初步的了解[237]。在当下,无论在溶液相还是固相中,Pu(Ⅶ)和Np(Ⅶ)均已不再罕见[98, 106];而二价锕系金属在不同相中的制备方法与理化性质也得到了较为深入的研究[90, 238-239]。
展望未来,锕系元素的非寻常氧化态化学仍有着广阔的发展前景。在气相,利用稀有气体基质的稳定作用[240],发展锕系金属-金属键物种的合成化学一直是令人兴奋的研究方向[34]。在固相,在稀土金属相关研究中发现的MX、M2X3、M5X8、M6X7、M7X10、M7X12(M=Y、La、Ce、Pr、Gd、Tb、Er、Ho、Lu)等卤化物簇有着重要的参考价值[90],为低价锕系金属固体化学的进一步发展提供了新的可能。对于溶液化学,离子液体由于其宽阔、可调的电化学窗口而成为发展锕系元素氧化还原化学的潜力介质[241]。曾有关于U(Ⅳ)、Th(Ⅳ)化合物在离子液体中的电化学还原的报道,但认为其过程分别为U(Ⅳ)→U(Ⅲ)→U(0)、Th(Ⅳ)→Th(0),并未经历U(Ⅱ)或Th(Ⅲ)、Th(Ⅱ)的形成[242-243]。对于后锕系元素而言,化学家们现在最主要的目标是,通过一系列对比研究,揭示结晶化合物中金属中心的基态电子构型,由此反映后锕系元素周期性的转变。在此基础上,比较与探索锕系元素和同样由正三价主导的镧系金属在成键性质和化学行为上的差异[151, 209, 244],从而实现核燃料后处理过程中镧-锕分离等关键过程的优化。
综上,锕系元素的非寻常氧化态研究无疑是当前锕系元素配位化学中最富生机、最具潜力、同时也最具挑战的领域之一。合成化学在这一领域的发展中起到了关键性的作用,但仍需其他方向上的协作才能取得更大进展。尤其是对于超钚元素来说,核素生产能力的提升[245-246]、传统测试方法的改良以及新表征技术的引入[209],是其配位化学研究在最近几年接连取得突破的先决条件。比如,2000年后,核素249Bk(T1/2=320 d)实现了毫克级的批量制备,这一技术突破极大地加速了锫的化学研究进展。又如,Galbis等[247]在2010年首次将扩展的X射线吸收精细结构(EXAFS)谱学方法与蒙特卡罗模拟相结合,解决了Cf3+水合离子结构的分辨问题。这推动了EXAFS在超钚元素溶液化学研究中的大量应用[244]。而结合磁圆二色谱(MCD)、电子顺磁共振(EPR)、超导量子干涉仪(SQUID)等表征手段,也将极大促进对锕系元素非寻常氧化态的电子结构、磁学特征的进一步了解。未来,锕系元素非寻常氧化态的研究将不仅限于基础研究,也将在诸多应用领域,如核燃料后处理、单分子磁体等方面展现巨大潜力[248]。通过更广泛、更深入的交流与合作,我们期待并坚信中国学者和中国团队会在锕系元素的非寻常氧化态化学和相关学科交叉领域做出更多的贡献。
猜你喜欢
材料保护(2022年2期)2022-12-07
学校教育研究(2021年20期)2021-12-14
中学生数理化(高中版.高考理化)(2021年10期)2021-12-06
防爆电机(2020年4期)2020-12-14
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21
农家科技中旬版(2019年3期)2019-07-08
中学化学(2019年2期)2019-07-08
广东教育·高中(2017年9期)2017-09-27
中文信息(2016年5期)2016-05-31
环球人物(2015年22期)2015-09-10
