
盐酸西那卡塞中间体有关物质的制备及纯化
2020-12-28 06:53龙玺国郑建东徐琳寓王崇益马玉恒
广州化学 2020年6期
王 栋, 龙玺国, 郑建东, 徐琳寓, 王崇益, 马玉恒*
(1. 滁州学院 材料与化学工程学院,安徽 滁州 239000;2. 江苏润安制药有限公司 生产技术部,江苏 淮安 223001)
盐酸西那卡塞(Cinacalcet)是一种新型口服拟钙剂,由美国NPS Pharmaceuticals公司开发。1977年,NPS制药公司将该产品转让给安进公司,并于2004年在美国获得上市许可,共有30 mg、60 mg、90 mg三个规格,商品名为Sensipar[1];之后在加拿大、欧盟、澳大利亚以及新西兰也获准上市。
1999年,协和发酵麒麟株式会社在日本进行了西那卡塞以治疗维持性透析患者的继发性甲状旁腺功能亢进为适应症的开发[2],并于2007年12月取得上市许可,规格为25 mg、75 mg,商品名为盖平/REGPARA;2018年,盖平片获批在中国上市,并被选为仿制药参比制剂。
通过查阅文献[3-6],西那卡塞可分别由相应的酰胺(XNKS-02)、烯烃(X2-a)、炔烃(X2-b)还原得到,其结构式如图1所示。

图1 酰胺(XNKS-02)、烯烃(X2-a)、炔烃(X2-b)结构式
其中XNKS-02是合成盐酸西那卡塞的重要中间体,WO2007127445A2报道了3-(3-三氟甲基苯基)丙酸及其衍生物(混合酸酐、酰卤)(X1)与 (R)-1-(1-萘基)乙胺(SM2)通过缩合反应得到的西那卡塞中间体(XNKS-02)[7]。该方法中用到了强腐蚀性,毒性强的酰卤试剂,为避开酰卤试剂,本文使用N,Nʼ-羰基二咪唑(CDI)作为缩合试剂,完成该步骤反应,方程式如图2所示。

图2 以CDI为缩合试剂合成XNKS-02
实验中发现,该步化学收率低于理论值,且中间体纯度无法达到内控标准,直接影响终产品的纯度,因此对反应过程和机理进行了研究。发现原料SM2除了参与合成中间体XNKS-02的反应外,在CDI的作用下还会发生自身缩合得到脲XNKS-02-P。
1 实验
1.1 主要仪器
IKA C-MAC-HS7磁力搅拌器;EYELA N-1300V-WB旋转蒸发仪;Biotage ISO-PSV制备色谱仪;Thermo Fisher U3000高效液相色谱仪;Bruker AV300型(300 Hz)核磁共振仪。
1.2 1,3-双[(R)-1-(萘-1-基)乙基]脲(XNKS-02-P’)的合成
将5.0 g (R)-(+)-1-(1-萘基)乙胺(0.029 mol)加入至50 mL三口瓶中,再加入25 mL二氯甲烷,搅拌溶清后,分批次加入2.37 g(0.015 mol)N,Nʼ-羰基二咪唑,20~30℃搅拌反应2 h,TLC(DCM/MEOH=10∶1)确认萘乙胺反应完全。体系中加入10 mL 1 mol/L盐酸水溶液,搅拌20 min,分层后收集有机相,再用10 mL水洗涤2次,经无水NaSO4干燥后过滤,滤液浓缩至干,得到棕色油状液体4.4 g,即XNKS-02-P’。
2 结果与讨论
2.1 合成路线设计
检索发现[8-10],已公开的关于化合物XNKS-02-P的制备方法具体如下:
Seung Hyo Kim在Org. Lett.发表了用钌螯合物作为催化剂,实现了胺类化合物在甲醇中直接缩合;方程式如图3所示。
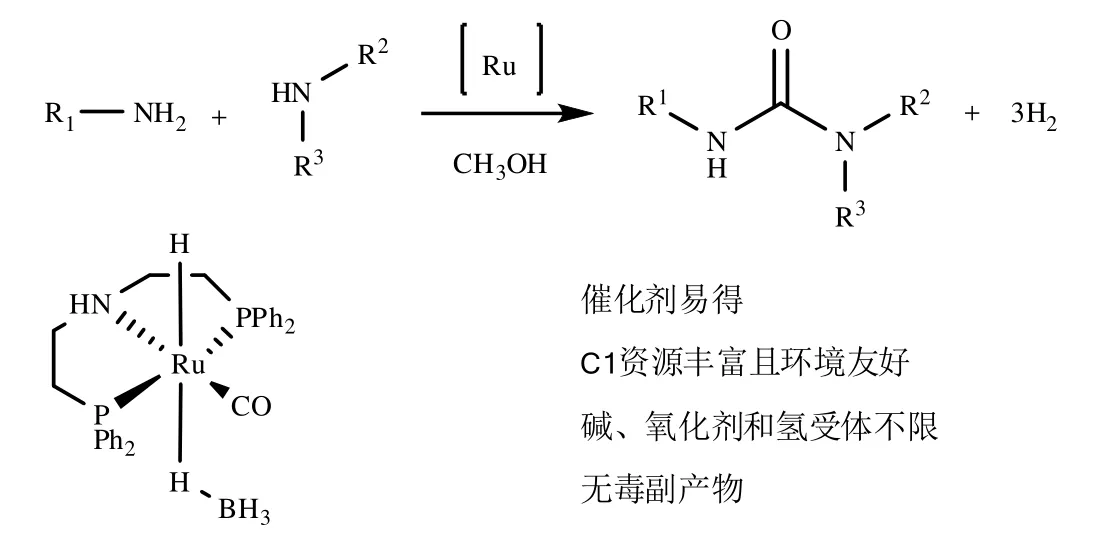
图3 胺类化合物在甲醇中直接缩合
Ji Hoon Park等人在Advanced Synthesis and Catalysis发表了在钴/铑杂比金属纳米颗粒的催化作用下,成功地实现了脂肪族和芳香型初级胺对脲的合成;方程式具体如图4所示。

图4 脂肪族和芳香型初级胺对脲的合成
上述两条路线中,催化剂分别为钌螯合物和钴/铑杂比金属纳米颗粒,其都属于稀有金属催化剂,价格昂贵,实际生产中还有重金属催化剂回收处理等问题。
B. M. Choudary在Synthetic Communications介绍了用异源化催化剂蒙脱石-双吡啶钯乙酸盐合成N,Nʼ-二取代氨基脲的方法,该方法中的催化剂需要定向合成,反应操作复杂,另外钯也是贵金属,同样也有成本高、回收处理困难等问题;
Balʼon, Ya在Journal of Organic Chemistry USSR发表的脲的合成中使用到了光气,光气毒性非常高,化学性质不稳定,遇水后有强烈的腐蚀性。
针对现有技术缺点,本文设计了一条反应温和、环境友好、成本低廉的化合物 XNKS-02-P’的合成路线,反应方程式见图5。

图5 由(R) - (+)-1- (1-萘基)乙胺与CDI在二氯甲烷中合成XNKS-02-P’
合成步骤具体如下:
1)将(R)-(+)-1-(1-萘基)乙胺、N,Nʼ-羰基二咪唑加入溶剂中,室温搅拌反应至萘乙胺反应完全;
2)体系用1 mol/L盐酸水溶液洗涤,静置分层后收集有机相,再水洗2次,干燥后,将溶剂浓缩至干,得到粗品;
2.2 1,3-双[(R)-1-(萘-1-基)乙基]脲(XNKS-02-P’)的分离
色谱柱:SNAP Ultra 100 g;流速:50 mL/min;紫外收集波长:254 nm;收集阈值:40 mAU。
取上述粗品4.4 g溶于20 mL二氯甲烷中,再加入4 g硅胶,搅拌均匀后浓缩至干得到载样硅胶,将其装入上样杯(FLASH Empty Column 12 g),按表1梯度洗脱(流动相A:正己烷,流动相B:乙酸乙酯)。

表1 梯度洗脱条件
收集XNKS-02-P’洗脱液(保留时间为11 min处对应的洗脱液),浓缩至干后真空干燥,得到1.32 g类白色固体即为XNKS-02-P’,纯度采用HPLC法,在215 nm检测,按面积归一化计算为95.62%。
另一方面,本文还提供了化合物XNKS-02-P’的高效液相色谱HPLC的检测方法,具体如下:
色谱柱:InfinityLab Poroshell 120 PFP(4.6×100 mm, 2.7 mm);流动相A:0.5%TFA水溶液;流动相B:0.3%TFA甲醇;流速:1.0 mL/min;进样量:10 μL;进样盘温度:5℃;柱温:35℃;检测波长:215 nm;梯度洗脱程序如表2所示。

表2 梯度洗脱条件
稀释剂:甲醇-水(1∶1);供试品浓度: 0.5 mg/mL。
经过HPLC定位比对,发现XNKS-02-P’的保留时间(保留时间为28.9 min)与合成中间体XNKS-02时产生的杂质XNKS-02-P的保留时间(保留时间为29.8 min)一致(详见图6、图7),从而确定了合成中间体XNKS-02时产生的杂质XNKS-02-P即为XNKS-02-P’。

图6 杂质XNKS-02-P’的HPLC光谱图

图7 XNKS-02的HPLC光谱图
2.3 1,3-双[(R)-1-(萘-1-基)乙基]脲(XNKS-02-P’)的表征
XNKS-02-P’:1H-NMR(DMSO-D6,300 MHz):d8.135 2~8.162 0(d, 2H), 7.923 4~7.954 4(m, 2H),7.789 7~7.836 1(m, 2H), 7.469 6~7.591 3(m, 8H), 6.414 5~6.441 5(d, 2H), 5.513 2~5.607 7(m, 2H),1.429 5~1.448 7(d, 6H);13C-NMR(CDCl3,300 MHz):d156.358, 141.208, 133.370, 130.286, 128.528, 127.006,125.983, 125.465, 125.406, 123.170, 121.875, 44.612, 22.436;MS:[M-H]-=367.2(详见图 8、图 9)。

图8 XNKS-02-P’的1H-NMR谱图

图9 XNKS-02-P’的13C-NMR谱图
3 结论
本文的意义在于证明了盐酸西那卡塞CDI缩合法合成中间体XNKS-02时产生特定杂质XNKS-02-P的机制,并且按照该机理成功制备以及分离纯化该杂质,随后进行了结构确证;另外,本文为大量获得该杂质,提供了一种新颖的、反应条件温和的、环境友好的合成方法,该方法成本低廉,产品纯度满足要求,可以作为定量标准品用于对中间体 XNKS-02的生产中控和成品检测,从而对原料药生产的质量把控起到重要作用。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
中学生数理化·中考版(2021年10期)2021-11-22
汕头大学学报(自然科学版)(2020年4期)2020-12-14
小哥白尼(趣味科学)(2020年5期)2020-05-22
有机氟工业(2019年2期)2019-08-12
妈妈宝宝(2018年9期)2018-12-05
中成药(2017年5期)2017-06-13
化工生产与技术(2016年5期)2016-03-13
股市动态分析(2015年12期)2015-09-10
心脑血管病防治(2011年3期)2011-09-15
