
罕见病相关体外诊断试剂的临床试验要求
2020-12-25 06:12何静云
国际检验医学杂志 2020年24期
徐 超,何静云,方 丽,李 冉
(国家药品监督管理局医疗器械技术审评中心,北京 100081)
罕见病的诊断与治疗是现代医疗需解决的问题,相关体外诊断试剂在该类疾病的诊疗过程中扮演重要角色。根据《体外诊断试剂注册管理办法》(国家食品药品监督管理总局令第5号)[1]要求,罕见病相关体外诊断试剂申请注册需进行临床评价,临床评价的证据来源主要为临床文献资料、临床经验数据、临床试验等。因罕见病相关体外诊断试剂一般存在风险较高,无已上市同类产品或已上市同类产品较少等特点,临床评价的主要方式为临床试验。
1 概 述
罕见病发病率低是该类疾病的重要特征,因此,相关产品临床试验过程中难以获得足够的病例数量,临床试验存在困难,生产企业对该类产品的研发、生产及注册申报缺乏积极性。为了解决相关问题,美国食品药品监督管理局(FDA)设立了人道主义豁免(HDE)上市前审批程序[2],该程序的适用标准为年度使用该器械的患者少于8 000例,符合此标准的医疗器械为人道主义豁免器械(HUD),此类产品在上市前评价过程中可适当减少临床试验病例数或免于进行临床试验。但是进入HDE程序的医疗器械并非均是罕见病防治产品,同时,有些用于罕见病防治的医疗器械上市也可能不适用于此程序。
我国药品监管部门一直重视罕见病相关产品的开发,中共中央办公厅、国务院办公厅发布的《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》[厅字(2017)42号][3]明确提出针对罕见病相关产品,在评价过程中可适当考虑减免临床试验。根据上述文件要求,为了鼓励生产企业对罕见病防治医疗器械的研发、生产,国家药品监督管理局发布了《国家药品监督管理局关于发布用于罕见病防治医疗器械注册审查指导原则的通告》(2018年第101号)[4](以下简称《指导原则》),《指导原则》以患者受益为出发点,科学解决用于罕见病防治医疗器械的临床评价难点,合理减免临床试验,以附带条件批准方式促进该类产品尽快用于临床,使罕见病患者受益。体外诊断试剂作为医疗器械的一部分,同样适用于《指导原则》,基于体外诊断试剂的特殊性,其临床研究与其他医疗器械临床研究存在差异,如何科学合理地设计相关产品临床试验一直是困扰行业的难题,也是急需解决的问题。国家药品监督管理局依据《指导原则》批准了水通道蛋白抗体(AQP4 Ab)检测试剂盒(酶联免疫法)[5],该产品的批准也为相关体外诊断试剂的试验确认提供参考。
2 适用范围
体外诊断试剂是否属于防治罕见病相关产品,应依据《指导原则》《关于公布第一批罕见病目录的通知》[国卫医发(2018)10号][6]及《国家卫生健康委办公厅关于印发罕见病诊疗指南(2019年版)的通知》[国卫办医函(2019)198号][7](以下简称《罕见病诊疗指南》)等文件判定。如申报产品临床适用证为第一批罕见病目录中的疾病,且依据《罕见病诊疗指南》该疾病的诊疗流程中需进行申报产品对应的检测项目的检测,则该产品可认定为防治罕见病相关产品。对于申报产品检测项目为新研发的生物标志物,应明确产品预期用途及其与相关罕见病诊疗的关系,从而判定其是否属于防治罕见病相关产品[8]。
案例:判定AQP4 Ab检测试剂盒(酶联免疫法)是否属于防治罕见病相关产品。首先,该产品临床适应证仅为视神经脊髓炎,该疾病属于第一批罕见病目录中的疾病;其次,根据《罕见病诊疗指南》要求,血清AQP4 Ab水平是辅助诊断视神经脊髓炎的一个检测项目,临床中AQP4 Ab检测与脊髓MRI检测、视觉诱发电位检查、光学相干光断层扫描仪检查(OCT检查)共同形成视神经脊髓炎的诊断与鉴别诊断标准。综上,该产品属于防治罕见病相关产品[8]。
3 预期用途
体外诊断试剂预期用途与产品的临床试验不可分割,临床试验的目标在于证明体外诊断试剂能够满足预期用途要求,并确定产品的适用人群及适应证。罕见病相关体外诊断试剂预期用途主要分为疾病的辅助诊断与疾病的筛查两类。
疾病诊断是通过症状、体征及各种检查结果来识别那些患有某种疾病或者具有某种症状的患者,同时能够排除那些未患该种疾病或不存在某种状态受试者的过程[9]。疾病辅助诊断类产品通过体外检测人体标本中的标志物,同时综合其他信息共同实现疾病诊断,如:AQP4 Ab检测试剂盒(酶联免疫法),通过检测血清AQP4-IgG结合MRI、视觉诱发电位、OCT检查等其他检查共同诊断患者是否为视神经脊髓炎,则该产品临床预期用途为视神经脊髓炎的辅助诊断。
疾病筛查是运用快速简便的试验、检查或其他方法将健康人群中那些可能有疾病或有缺陷,但表观健康的个体,同那些可能无疾病者鉴别开,它是从健康人群中早期发现可疑患者的一种措施,不是对疾病做出诊断[10]。用于罕见病筛查的产品如苯丙氨酸检测试剂盒(荧光法),该产品通过检测采集在滤纸上的新生儿全血样品中的苯丙氨酸水平,从而对新生儿苯丙酮尿症进行筛查,检测结果阳性需召回复查,复查仍阳性则需进行鉴别诊断。该产品临床预期用途为疾病筛查。
产品预期用途的确定与产品临床应用及临床试验息息相关,是产品研发早期需确定的内容。相关产品生产研发单位应依据相关产品诊疗方案、诊疗指南、诊断标准及专家共识确定产品预期用途,同时针对已确定的预期用途开展合理的临床试验。
4 临床试验要求
与罕见病相关治疗类产品不同,体外诊断试剂适用人群除罕见病患者以外,还包括疑似罕见病患者、健康者等其他人员。产品预期使用情况应为真正的患者可以被准确地诊断,从而采取合理的干预措施;同时,非该疾病的患者不会因为检测结果的假阳性而接受不必要的处置。为了验证产品的安全有效,产品应进行合理的临床试验。
4.1临床试验设计 体外诊断试剂临床试验一般采用观察性研究,研究设计一般采用同步盲法比较考核试剂检测结果与已上市同类产品和/或临床诊断标准[11]之间的一致性。该类研究主要涉及横断面研究和纵向数据研究两类。
横断面研究即评价单一时间点采集标本的检测结果与临床参考标准(或其他方法)判定结果的一致性。如:AQP4 Ab检测试剂盒(酶联免疫法)临床试验设计。该研究采用考核试剂对来自临床试验入组病例者的血清中AQP4 Ab水平进行检测,根据检测结果将入组病例分为检测结果阳性人群与检测结果阴性人群。同时,入组病例根据临床诊断标准同样可分为“金标准”确定“有该病”人群及“金标准”确定“无该病”人群,将考核试剂检测结果与临床诊断结果进行比对,以考察考核试剂的临床性能。该设计为典型的横断面研究。
纵向数据研究即需要多个时间点采集标本的检测结果,并结合病例临床状态才能确定评价产品临床性能。例如,某些用于治疗监测的体外诊断试剂,在临床试验中应对受试者及其标本中的被测物进行治疗前后多个时间点的观测,以证明被测物检测结果的变化与病情发展、治疗效果的相关性。目前鲜见用于罕见病治疗监测及预后评估的产品上市。
4.2对比方法选择 体外诊断试剂临床试验对比方法的选择至关重要,直接关系到产品临床试验的结果。罕见病相关体外诊断试剂临床试验对比方法可选择疾病临床参考标准或已上市同类产品等,以下针对两种对比方法的选择进行介绍。
罕见病相关体外诊断试剂已上市产品较少,针对用于某一种罕见病辅助诊断的产品,其临床试验过程中,往往很难获得已上市同类产品作为对比产品,该类产品进行临床试验对比方法可选择临床诊断“金标准”。“金标准”是指在现有条件下,公认的、可靠的、权威的诊断方法。临床上常用的“金标准”有组织病理学检查、影像学检查、病原体分离培养鉴定、长期随访所得的结论及临床常用的其他确认方法等[11],罕见病相关诊断的“金标准”应来自《罕见病诊疗指南》。一般而言,产品与临床诊断“金标准”进行比对的同时,还应考虑采用其他方法验证产品临床检测的准确性。
除疾病诊断“金标准”外,另一种临床参考标准为针对某些标志物临床公认的检测方法。采用此临床参考标准作为对比,可对产品临床标本检测的准确性进行评估。如基因检测的产品,临床对比方法可选择基因测序作为对比方法;某些激素及小分子化合物检测的产品,临床可采用公认的色谱技术作为对比方法。需要注意的是,为了更充分地评价产品临床性能,在产品与此类临床参考标准对比的基础上,还需考虑产品与临床诊断“金标准”的一致性。
针对已有同类产品上市的罕见病相关体外诊断试剂,其临床试验可选择已上市同类产品作为对比。临床试验为评估待考核试剂与已上市同类产品一致性。目前针对多个罕见病已有相关产品上市,如针对肝豆状核变性辅助诊断的铜蓝蛋白检测试剂,在国家药品监督管理局网站数据查询系统检索,已有11个同类产品上市。
4.3入组人群 临床试验入组人群应来自产品预期用途所声称的适用人群(目标人群),如具有某种症状、体征、生理、病理状态或某种流行病学背景等情况的人。非目标人群入组可能引入受试者选择偏倚,导致临床试验结果不能反映产品的真实情况。同时在人群的入组过程中,应重点考虑入组部分可能对检测结果产生干扰的病例及易与目标状态相混淆的病例等。
案例:AQP4 Ab检测试剂盒(酶联免疫法),其预期用途为视神经脊髓炎的辅助诊断,其临床试验入组人群应为临床表现可能为视神经脊髓炎的人群,临床表现包括:视神经炎、急性脊髓炎、极后区综合征、急性脑干综合征、症状性睡眠发作或急性间脑临床综合征伴视神经脊髓炎谱系疾病典型的间脑MRI病灶、症状性大脑综合征伴视神经脊髓炎谱系疾病典型的脑部病变等[12]。为了充分评估产品临床性能,还应入组部分干扰病例,如其他中枢神经系统脱髓鞘病(多发性硬化症、急性播散性脑脊髓炎、假瘤型脱髓鞘病等),血管性疾病(缺血性视神经病、脊髓血管畸形、亚急性坏死性脊髓病等),感染性疾病(结核、艾滋病等),代谢中毒性疾病(中毒性视神经病、亚急性联合变性等),遗传性疾病(Leber视神经病等),肿瘤及副肿瘤相关疾病(脊髓胶质瘤、室管膜瘤等),以及临床其他需要与视神经脊髓炎进行鉴别诊断的病例。
临床试验入组人群的方式有两种,第一种为入组人群分为两组:一组是用“金标准”确定为有某病的病例组,另一组是经“金标准”确定或有临床证据证实无该病的患者或健康人群作为对照组,将两组病例混合、编盲开展临床试验,此种方式入组人群临床试验模式见图1;第二种为入组人群为某一疾病的疑似病例,将所有入组病例进行编盲开展临床试验,进行考核产品与临床诊断标准(或已上市同类产品)的比对,此种方式入组人群的临床试验模式见图2。第一种人群入组方式,在病例收集过程中能够更有针对性地收集阳性病例,但是各种病例的构成与临床真实情况相差较大;第二种人群入组方式,不能有针对性地收集阳性病例,阳性病例数量取决于阳性病例在适用人群中的占比,但各种病例的构成与临床真实情况相符。有研究表明,以第一种方式入组人群的临床试验会高估产品的临床诊断性能[13]。

图1 第一种入组人群模式

图2 第二种入组人群模式
4.4病例数量 符合统计学要求的病例数量是保证体外诊断试剂临床性能得到准确评价的必要条件。对罕见病而言,发病率低、病例数量少是相关体外诊断试剂临床研究的困难,《指导原则》从几个方面解决了难题:(1)临床试验要求能够证明产品潜在的安全有效性即可;(2)允许产品上市后继续进行产品临床应用情况的研究。产品上市前与上市后数据综合分析,评价产品临床性能。以下针对罕见病诊断或辅助诊断产品及罕见病筛查产品临床试验病例数要求进行探讨。
4.4.1罕见病诊断或辅助诊断产品 针对适用范围为罕见病诊断或辅助诊断的产品,其适用人群除了相关罕见病患者外,还包括部分非罕见病患者。临床试验入组的病例数量与产品评价指标相关。针对产品临床试验中所需阳性病例的数量,应依据产品临床灵敏度点估计值、疾病流行病学特征及临床试验机构条件等因素确定,阳性病例可不满足统计学要求,其中产品临床灵敏度点估计值为产品潜在临床性能的重要评价标准。如某产品临床试验过程中根据流行病学特征和医疗机构的条件入组了n例阳性病例,针对已入组的阳性病例进行产品临床灵敏度点估计,如满足临床需求,则可认为产品潜在的临床性能被确认。
罕见病相关产品临床试验过程中所需阴性病例,大部分情况下该部分病例并不罕见,因此,应根据产品临床特异度确定临床试验中需入组的阴性病例数量,阴性病例应重点考虑对该疾病诊断可能存在干扰的病例。阴性病例应满足统计学要求,针对体外诊断试剂产品,常见的临床试验标本量估算方法有两种。
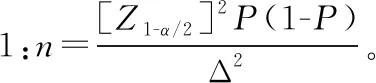
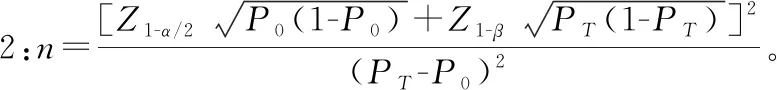
案例:AQP4 Ab检测试剂盒(酶联免疫法)临床试验,申报产品与临床诊断“金标准”进行对比研究过程中,综合考虑既往相关技术在临床应用的情况、临床可接受的标准及产品性能等因素,确定产品临床灵敏度不低于85%,特异度不低于95%,允许误差临床可接受的标准为0.05,Ⅰ类错误概率α设定为双侧0.05,采用公式1进行标本量估算,得出符合统计学要求的阳性病例为196例,阴性病例为73例。根据疾病流行情况及临床试验机构标本留存情况,入组阳性病例55例,阳性病例不满足统计学要求,但是针对55例病例进行统计分析得出产品灵敏度为89%,能够证明产品潜在的临床性能;阴性病例入组119例,满足统计学要求。因此该产品临床试验入组174例病例,能够满足评价产品潜在安全有效性要求,需在产品上市后继续收集临床使用数据。
4.4.2罕见病筛查产品 罕见病筛查是体外诊断试剂的重要临床应用场景。针对适用范围为罕见病筛查的产品,临床试验入组人群应为该产品目标适用人群,如健康人群或高风险人群,为了评价产品临床筛查性能,临床试验应依据疾病发病率保证临床试验过程中至少有真阳性病例筛出,临床试验只有满足真阳性病例筛出的标准,才能初步评估产品的临床灵敏度及阳性预测值,才能证明产品潜在的临床价值。此外,临床试验可包含部分已确诊的病例,进行回顾性研究,以补充评价产品临床标本检测性能。
4.5统计分析 罕见病相关体外诊断试剂临床试验统计分析一般采用绘制四格表的方式[14],评价其临床诊断性能。依据疾病诊断“金标准”或已上市同类产品将入组人群分为“阳性”和“阴性”,同时,依据考核试剂检测结果将入组人群分为“阳性”和“阴性”,绘制四格表见表1。用于罕见病诊断或辅助诊断的产品,其统计指标应包括临床灵敏度(阳性符合率)、临床特异度(阴性符合率),其中临床特异度(阴性符合率)应计算相应置信区间。用于罕见病筛查的产品,其统计指标应包括临床灵敏度、临床特异度、阳性预测值、阴性预测值、阳性似然比、阴性似然比等,其中临床特异度、阴性预测值等指标应计算相应置信区间。结合表1内容具体统计指标的描述及计算公式详见表2。

表1 诊断试验四格表

表2 相关统计指标描述及计算公式

续表2 相关统计指标描述及计算公式
5 总 结
从国家药品监督管理局官方网站查询得知,我国目前已批准部分罕见病相关体外诊断试剂产品,如用于肝豆状核变性辅助诊断的铜蓝蛋白检测试剂盒;用于21-羟化酶缺乏症辅助诊断的血清17羟孕酮(17-OHP)检测试剂盒;用于视神经脊髓炎辅助诊断的血清AQP4-IgG检测试剂盒;用于新生儿苯丙酮尿症筛查的苯丙氨酸检测试剂等,产品预期用途涵盖辅助诊断与筛查等多个方面。罕见病为多个疾病的统称,与数量较多的疾病种类相比,我国批准的产品数量较少。
本文介绍的关于罕见病相关体外诊断试剂临床试验要求基于当前认知和相关产品临床评价经验。随着产品评价技术的不断进步,一些新的临床评价思路及新的统计学方法可能在相关产品的评价中被更加科学合理地应用。如针对罕见病发病率低、临床试验病例收集困难的问题,真实世界数据的应用[15]、临床试验适应性设计[16]可能为产品临床试验困难提供新的解决思路。贝叶斯分析等新的统计学方法也可能在罕见病相关体外诊断试剂临床试验过程中发挥作用。关于该类产品科学合理的临床评价是产品开发者与监管部门共同研究的课题。
包括我国在内的很多国家存在罕见病患者诊断困难的问题,超过30%的患者要经过5~10名医师诊断,确诊周期可长达5~30年[17]。罕见病相关体外诊断试剂的开发是解决我国罕见病患者诊断困难的一个重要途径。针对该类疾病的特点,相关产品在临床试验过程中存在对比方法难以确定、病例数少等问题,申请人在产品研发验证过程中应重点进行产品风险评估。在临床试验过程中,应依据该类疾病患者临床诊疗现状,科学、合理地设计临床试验,在临床试验阳性病例数不满足统计学要求的情况下,论证产品临床应用潜在的安全有效性,促进产品尽快上市,满足临床需求。
猜你喜欢
现代仪器与医疗(2021年1期)2021-06-09
恋爱婚姻家庭(2020年27期)2020-10-09
基层中医药(2020年5期)2020-09-11
百花洲(2018年1期)2018-02-07
中国资源综合利用(2017年4期)2018-01-22
瞭望东方周刊(2017年45期)2017-12-08
现代检验医学杂志(2016年1期)2016-11-12
浙江人大(2014年6期)2014-03-20
中国合理用药探索(2012年2期)2012-03-20
中国合理用药探索(2011年9期)2011-03-20
