
新型3,4,5-三取代噁唑烷酮类化合物的合成
2020-12-25 01:11樊陈莉冯光峰张世文
安徽师范大学学报(自然科学版) 2020年6期
樊陈莉,张 强,冯光峰, 张世文
(1.芜湖职业技术学院 材料工程学院,安徽 芜湖 241003;2.安徽师范大学 化学与材料科学学院,安徽 芜湖 241002)
噁唑烷酮是一类重要的杂环骨架,因其具有重要的生物活性及材料特性而广泛应用于医药、材料等领域[1-2],同时噁唑烷酮类化合物也是一类非常重要的有机合成中间体,可以发生多种化学转化,常被应用于一些抗菌剂、除草剂、聚合物改性和新型高性能聚氨酯的合成等[3-6]。因此,寻找噁唑烷酮母环骨架简便高效的合成方法,是许多有机化学家和药物学家所关注的问题,也是近年来有机合成领域研究的热点之一。
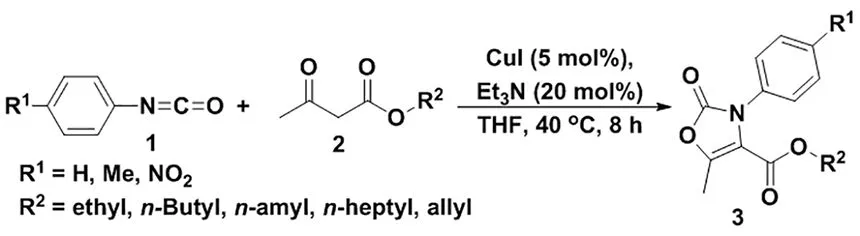
图1 三取代噁唑烷酮类化合物的设计合成Scheme 1 Synthesis of 3,4,5-trisubstituted oxazol-2(3H)-one derivatives
传统合成噁唑烷酮的方法是以光气或光气的类似物为原料进行环化,这些试剂不仅具有较大的毒性,而且实验操作繁琐,使用大量的溶剂,既造成了资源的浪费,又严重的污染环境。随后,一些合成噁唑烷酮环的新方法陆续被开发[7],如分子内氧烷基化[8]、多组分合成[9]、串联反应[10]等。噁唑烷酮类化合物还是一种重要的抗菌药物,随着耐药菌的不断出现,药物研究学者们越来越重视此类物质合成方法的研究。因此,发展简单、快速合成出结构新颖、具有生物活性的噁唑烷酮衍生物的方法将成为有机合成领域研究的热门课题。在课题组前期研究的基础上,本文发展了一种CuI催化的异氰酸酯与β-酮酯的[3+2]环加成反应合成得到一种结构新颖的多取代噁唑烷酮类化合物,设计合成路线如图1所示。
1 实验部分
1.1 仪器与试剂
BRUKER-AV-300型核磁共振仪(300MHz,CDCl3为溶剂,TMS为内标);X-4数字显示显微熔点测定仪(北京泰克仪器有限公司,温度计未经校正)。
实验所用溶剂和试剂均为分析纯(可直接使用)。
1.2 噁唑烷酮类化合物3a~3h的合成
取一干燥的双颈瓶,向其中加入CuI(0.05mmol),并抽真空,充氮气。随后在氮气保护下依次加入异腈酸酯1(1mmol),β-酮酯2(3mmol)和四氢呋喃(3mL),最后在搅拌下利用恒压滴液漏斗向反应液中逐滴加入三乙胺(0.2mmol),滴加完毕后将反应液加热至40℃反应8小时。反应结束后,将反应体系冷却至室温,减压蒸去溶剂,水洗,用乙酸乙酯做萃取剂,萃取3次,每次10mL,收集有机相,无水硫酸钠干燥,过滤,用旋转蒸发仪蒸去大量溶剂,剩余物用柱色谱分离。[洗脱剂:V(石油醚)/V(乙酸乙酯)=10/1至15/1(v/v)]分离得到噁唑烷酮类化合物3a~3h。
化合物3a: 白色固体,收率80%,m.p.91~92℃;1H NMR(CDCl3,300MHz)δ:7.55(d,J=8.1Hz,2H,C6H5),7.34(t,J=7.8Hz,2H,C6H5),7.17(t,1H,J=7.2Hz,C6H5),4.32(q,J=6.9Hz,2H,OCH2CH3),2.49(s,3H,CH3),1.40(t,J=6.9Hz,3H,OCH2CH3);13C NMR(CDCl3,75MHz) δ:192.1,170.8,169.2,136.9,128.9,124.8,121.4,95.0,60.9,26.4,14.3;HR-MS(ESI-TOF)m/z:Calcd for C13H14NO4{[M+H+}248.092 3,found 248.092 8。
化合物3b:白色固体,收率81%,m.p.85~87℃;1H NMR(CDCl3,300MHz)δ:7.53(d,J=7.5Hz,2H,C6H5),7.33(t,J=7.2Hz,2H,C6H5),7.12(t,J=6.9Hz,1H,C6H5),4.19(t,J=6.0Hz,2H,OCH2CH2CH2CH3),2.44(s,3H,CH3),1.34~1.42(m,4H,OCH2CH2CH2CH3),0.92(t,J=6.0Hz,3H,OCH2CH2CH2CH3);13C NMR(CDCl3,75MHz)δ:192.0,1708,169.2,137.0,128.9,124.6,121.1,94.9,65.1,28.3,26.4,22.3,14.0;HR-MS(ESI-TOF)m/z:Calcd for C15H18NO4{[M+H+}276.123 6,found 276.123 1。
化合物3c:白色固体,收率81%,m.p.85~87℃;1H NMR(CDCl3,300MHz)δ:7.54(d,J=7.5Hz,2H,C6H5),7.35(t,J=7.2Hz,2H,C6H5),7.12(t,J=7.2Hz,1H,C6H5),4.19(t,J=6.9Hz,2H,OCH2CH2CH2CH2CH3),2.45(s,3H,CH3),1.32~1.43(m,6H,OCH2CH2CH2CH2CH3),0.92(t,J=6.9Hz,3H,OCH2CH2CH2CH2CH3);13C NMR(CDCl3,75MHz)δ:192.1,170.9,169.2,137.1,128.9,124.7,121.2,94.7,65.2,28.3,26.5,22.4,14.0;HR-MS(ESI-TOF)m/z:Calcd for C16H20NO4{[M+H+}290.139 2,found 290.139 6。
化合物3d:白色固体,收率75%,m.p.82~83℃;1H NMR(CDCl3,300MHz)δ:7.39(d,J=7.5Hz,2H,C6H5),7.33(t,J=7.5Hz,2H,C6H5),7.06(t,J=7.5Hz,1H,C6H5),4.17(t,J=6.9Hz,2H,OCH2CH2CH2CH2CH2CH2CH3),2.37(s,3H,CH3),1.59~1.75(m,10H,OCH2CH2CH2CH2CH2CH2CH3),0.88(t,J=6.9Hz,3H,OCH2CH2CH2CH2CH2CH2CH3);13C NMR(CDCl3,75MHz)δ:192.1,170.9,169.2,140.0,129.2,128.7,124.8,121.4,95.1,64.2,27.3,27,22.4,17.4,14.3,13.1;HR-MS(ESI-TOF)m/z:Calcd for C18H24NO4{[M+H+}318.170 5,found 318.170 2。

化合物3f: 白色固体,收率81%,m.p.115~116℃;1H NMR(CDCl3,300MHz)δ:7.96(d,J=7.5Hz,2H,C6H4),7.28(d,J=7.2Hz,2H,C6H4),4.19(q,J=6.9Hz,2H,OCH2CH3),2.47(s,3H,CH3C6H4),2.43(s,3H,CH3),1.29(t,J=6.3Hz,3H,OCH2CH3);13C NMR(CDCl3,75 MHZ)δ:166.0,164.3,163.9,144.7,130.1,129.3,126.2,110.3,60.2,29.7,21.7,18.2,14.2;HR-MS(ESI-TOF)m/z:Calcd for C14H16NO4{[M+H]+}262.107 9,found 262.107 3。
化合物3g: 白色固体,收率85%,m.p.108~109℃;1HNMR(CDCl3,300MHz)δ:7.97(d,J=8.1Hz,2H,C6H4),7.29(d,J=8.1Hz,2H,C6H4),4.31(t,J=6.3Hz,OCH2CH2CH2CH2CH2CH2CH3),4.13~4.17(m,2H,OCH2CH2CH2CH2CH2CH2CH3),2.49(s,3H,CH3C6H4),2.43(s,3H,CH3),1.67~1.77(m,4H,OCH2CH2CH2CH2CH2CH2CH3),1.31(m,4H,OCH2CH2CH2CH2CH2CH2CH3),0.90(t,J=6.9Hz,3H,OCH2CH2CH2CH2CH2CH2CH3);13C NMR(CDCl3,75 MHZ)δ:195.9,166.2,164.3,164.0,144.7,130.1,130.0,129.5,129.3,129.1,129.0,126.2,110.3,67.2,64.9,64.4,31.8,31.7,28.9,28.7,28.6,26.0,25.9,25.6,22.5,21.7,21.6,18.3,14.1;HR-MS(ESI-TOF)m/z:Calcd for C19H26NO4{[M+H]+}332.186 2,found 332.186 7。
化合物3h: 淡黄色固体,收率68%,m.p.119~120℃;1H NMR(CDCl3,300MHz)δ:8.25(d,J=8.7Hz,2H,C6H4),8.17(d,J=8.4Hz,2H,C6H4),4.13(q,J=6.9Hz,2H,OCH2CH3),2.41(s,3H,CH3),1.24(t,J=7.2Hz,3H,OCH2CH3);13C NMR(CDCl3,75 MHZ)δ:164.4,162.2,160.8,149.5,133.1,130.0,122.5,109.7,59.2,16.8,13.0;HR-MS(ESI-TOF)m/z:Calcd for C13H13N2O6{[M+H]+}293.077 4,found 293.077 0。
2 结果与讨论
首先以异氰酸酯(1a)和乙酰乙酸乙酯(2a)为模板底物在CuI(5mol%)和三乙胺(TEA,20mol%)催化下对实验条件进行了优化,主要考察了β-酮酯的用量、碱、溶剂、反应温度和反应时间的影响(表 1)。结果表明,当使用1倍当量β-酮酯时,改变不同的温度,也仅能以45%的收率得到目标产物3a(表 1,序号1~3)。进一步增加β-酮酯用量,目标产物的产率有明显提高(表1,序号4~6),当使用3倍当量的β-酮酯作为底物时,产率达到最高(表1,序号5)。随后考察了四氢呋喃(THF)、二氯甲烷(DCM)、乙腈(MeCN)、甲苯(toluene)、二甲基甲酰胺(DMF)等溶剂对反应的影响,结果显示四氢呋喃是最佳反应溶剂(表 1,序号3,7~10)。最后考察了不同碱(如吡啶、磷酸钾、碳酸钾)及反应时间的影响,发现该反应以三乙胺作碱在40℃下反应8小时产率最好(表1,序号11~15),并以此作为该反应的最优实验条件进行底物普适性研究。
在最优化实验条件下,对底物取代基的影响进行了探究(表2)。结果表明,各种不同取代基的β-酮酯均能与苯基异氰酸酯(2a)发生反应并以75%~81%的产率得到目标产物(表2,序号1~4),且酯基上烷基链的长度对反应没有明显的影响。值得一提的是,使用乙酰乙酸烯丙酯作为底物时,该反应也能很好的进行,并能以70%的产率得到相应的噁唑烷酮化合物3e(表2,序号5),且产物中含有的烯丙基可以实现多种化学转化,能够进一步丰富该类化合物结构类型。随后考察了异氰酸酯苯基上取代基的影响,结果表明当苯基异氰酸酯的对位上连有供电子基团(如—CH3)时有利于反应的进行,产物的产率比较高(表2,序号6,7),而当对位上连有强吸电子基团(如—NO2)时,异氰酸酯的反应活性较低,不利于反应的进行,仅能以68%的产率获得目标产物(表2,序号8)。遗憾的是,当在β-酮酯结构中引入苯基时,该反应不能发生(表2,序号9,10)。
根据文献报道[11]、反应底物及产物的结构等,对该反应提出了一种可能的反应机理(图2)。首先,β-酮酯发生烯醇异构化生成中间体A,随后烯醇中间体A在三乙胺作用下失去质子并与CuI配位得到中间体B,最后中间体B与苯基异氰酸酯发生[3+2]环加成反应得到噁唑烷酮类化合物3。

表1 实验条件的优化

表2 底物普适性探究
3 结论
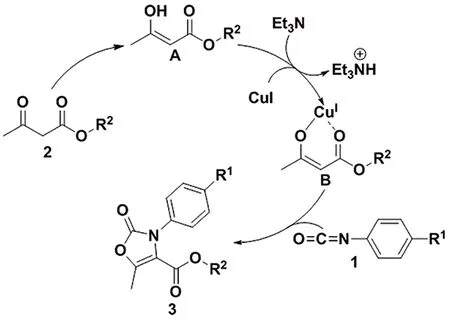
图2 可能的反应机理Scheme 2 Proposed mechanism
以廉价易得的CuI为催化剂,在温和条件下实现异氰酸酯和β-酮酯类化合物的[3+2]环加成反应,发展了一种结构新颖的多取代噁唑烷酮衍生物的合成方法。具有实验条件温和、后处理简单、反应选择性好等优点,为研究开发具有药物活性的噁唑烷酮类化合物的合成提供了一种经济、切实可行的方法。
猜你喜欢
当代化工研究(2022年19期)2022-11-04
纺织标准与质量(2022年2期)2022-07-12
农业与技术(2021年3期)2021-12-06
教育周报·教育论坛(2020年3期)2020-10-21
北京化工大学学报(自然科学版)(2020年2期)2020-06-22
宇航材料工艺(2020年1期)2020-03-26
科学大观园(2019年23期)2019-09-10
科技资讯(2018年16期)2018-10-26
科技信息·下旬刊(2018年8期)2018-10-21
科技资讯(2017年12期)2017-06-09
