
基于生物信息学的子宫内膜癌诊断标志物的筛选及免疫细胞浸润分析
2020-12-24 07:09阿说阿牛曲木金作伍秋红凉山州妇幼保健计划生育服务中心妇产科凉山615000
中国免疫学杂志 2020年22期
阿说阿牛 孙 丽 曲木金作 伍秋红 (凉山州妇幼保健计划生育服务中心妇产科,凉山 615000)
子宫内膜癌(endometrial carcinoma,EC)是常见的妇科恶性肿瘤之一[1]。由于人口老龄化和肥胖率上升,全球范围内EC的发病率和死亡率呈上升趋势[2]。目前,EC的早期诊断通常基于患者临床表现,如绝经后出血或某些肿瘤标志物的血清水平异常,而约15%的EC发生于无阴道出血的女性[3]。有研究报道了不同的血清学标志物在EC诊断中的作用,如癌胚抗原、碳水化合物抗原-125和碳水化合物抗原19-9,但其仅在20%~30%的EC患者中表达上调[3,4]。由于诊断不及时,EC患者往往失去了最佳治疗时机,导致肿瘤转移及术后复发的风险较高,预后较差[5,6]。因此,迫切需要寻找更可靠和前瞻性更强的诊断生物标志物以提高EC患者的生存率。越来越多的证据表明,肿瘤免疫细胞浸润与癌症的发生发展密切相关[7-9]。肿瘤中免疫细胞浸润的类型和比例与临床结果密切相关,其不仅对患者的生存具有预测价值,还可影响肿瘤治疗效果,因此有望成为药物靶标及临床生物标志物[10,11]。肿瘤相关中性粒细胞是免疫细胞的主要类型,可消除病原体并防止宿主受到微生物感染,且与乳腺癌和胃癌的预后相关[12-14]。此外,肿瘤相关巨噬细胞参与EC的侵袭性进展,可作为EC的潜在治疗靶标[15,16]。本研究从GEO和TCGA数据库下载EC的表达谱数据集,利用R软件鉴定差异表达基因(differentially expressed genes,DEGs),并对DEGs进行功能和通路富集分析[17,18]。构建蛋白质-蛋白质相互作用(protein-protein interaction,PPI)网络并选择关键(hub)基因,通过绘制ROC曲线对hub基因进行诊断价值分析,进而筛选出EC的诊断生物标志物。CIBERSORT是基于线性支持向量回归原理对免疫细胞亚型的表达矩阵进行去卷积的工具,可利用RNA-Seq的数据预估免疫细胞浸润情况[19]。本研究利用CIBERSORT对EC患者进行免疫细胞浸润分析,初步探究免疫细胞浸润在EC中的作用,以期为EC提供诊断生物标志物,并初步评估EC中免疫细胞浸润情况,为进一步研究EC的分子机制提供新思路。
1 材料与方法
1.1材料 通过GEO和TCGA数据库下载EC的表达谱数据。GSE115810数据集基于GPL96([HG-U133A] affymetrix human genome U133A Array)平台,包括3例子宫内膜样本和24例EC样本;GSE17025数据集基于GPL570([HG-U133_Plus_2] affymetrix human genome U133 plus 2.0 Array)平台,包括12例子宫内膜样本和91例EC样本;来源于TCGA数据库的数据集包括13例子宫内膜样本和174例EC样本[20]。
1.2方法
1.2.1数据预处理及DEGs鉴定 采用R软件的affy包读取GSE115810和GSE17025 2个数据集的原始数据,采用RMA算法进行背景校正、数据归一化处理[21]。在Bioconductor软件应用GPL96和GPL570平台所对应的基因注释文件对探针矩阵进行注释[22]。使用sva包去除批间差,并转化为探针表达矩阵,使用分位数-分位数(Q-Q)图可视化去除批间差效果,样本间校正效果则用二维PCA聚类图进行展示[23]。TCGA数据经校正后转化为CIBERSORT能够处理的表达矩阵。使用R语言limma包进行差异表达分析并输出DEGs[24]。DEGs满足adj.Pvalue<0.05及|log2FC|>1。通过Funrich软件对GEO和TCGA数据集的DEGs进行求交集,并使用ggplot2包绘制DEGs的火山图,直观展示DEGs的差异表达情况[25,26]。
1.2.2GO和KEGG通路富集分析 GO涵盖细胞组分、分子功能和生物过程3个生物学过程[27]。KEGG是从分子水平了解生物系统高层次功能的数据库[28]。为进一步分析DEGs的功能,使用R软件的clusterProfile包对DEGs进行GO和KEGG通路富集分析,以P<0.05为差异具有统计学意义[29]。
1.2.3GSEA分析 选择c2.cp.kegg.v6.0.symbols.gmt作为参考基因集进行CTSEA分析。截止标准设置为FDR<0.25且P<0.05,分析结果使用R软件进行可视化。
1.2.4PPI网络构建及hub基因选择 采用STRING数据库构建PPI网络,采用Cytoscape软件将PPI网络可视化[30,31]。利用cytoHubba插件选择最大相关标准中的前10个基因作为hub基因[32]。
1.2.5诊断生物标志物的筛选 ROC分析是把灵敏度和特异度结合起来综合评价诊断准确度或判别效果的一种方法。采用R语言的pROC包绘制ROC曲线评估10个hub基因的诊断价值,进而筛选出EC的诊断生物标志物[33]。
1.2.6免疫细胞浸润程度评估 利用corrplot包绘制相关性热图以可视化22种免疫细胞浸润的相关性[34];ggplot2包绘制小提琴图用于可视化22种免疫细胞浸润差异。
2 结果
2.1数据预处理及DEGs鉴定 GSE115810和GSE17025数据集基因表达矩阵数据去除批间差后的效果表明两组样本的批间差已经去除(图1)。合并后的矩阵标准化处理前后效果以二维PCA聚类图呈现(图2A、B),标准化处理后两组样本聚类更为明显,表明样本来源可靠。数据预处理后,利用R软件从GSE115810和GSE17025数据集中提取675个DEGs,从来源于TCGA的数据集中获得7 185个DEGs,重叠部分共有448个DEGs(图2C)。DEGs的火山图表明DEGs在EC中明显差异表达(图2D)。
2.2功能和通路富集分析 GO分析结果表明,DEGs的生物学过程变化显著富集于染色体分离、核分裂和纺锤体组织,细胞成分变化主要在着丝粒、细胞间桥和细胞外基质,分子功能变化主要富集于趋化因子活性和丝氨酸肽酶活性(图3A)。KEGG通路富集分析结果显示, 富集的通路主要与病毒蛋白与细胞因子和细胞因子受体的相互作用通路、趋化因子信号通路、TGF-β信号通路及细胞周期信号通路密切相关(图3B)。
GSEA分析结果显示,富集的通路除包括病毒蛋白与细胞因子和细胞因子受体的相互作用通路、趋化因子信号通路、 TGF-β信号通路和细胞周期信号通路外,还涉及HIF-1信号通路、IL-17信号通路及Wnt信号通路等(图4A)。其中CDC27、 E2F1和BUB1是细胞周期信号通路的重要参与者,VEGFA、AKT1和PIK3R2在HIF-1信号通路转导过程中具有重要作用(图4B)。

图1 GSE115810和GSE17025数据集去除批间差的密度图及Q-Q图Fig.1 Density and Q-Q map of difference between batches of GSE115810 and GSE17025 removed datasets
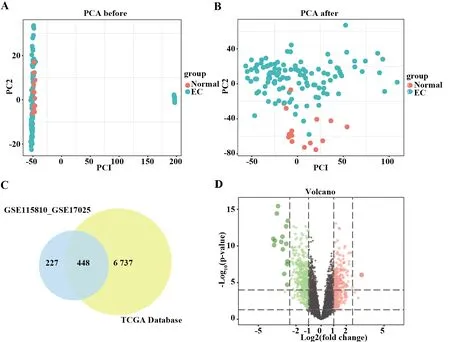
图2 二维PCA聚类图、韦恩图及火山图Fig.2 2D-PCA cluster map,venn diagram and volcano plotNote:A,B.2D-PCA cluster map of GEO database samples before and after correction;Red represents normal endometrial sample group,blue represents EC sample group; abscissa and ordinate represent clustering dimension reduction surface;C.GEO and TCGA datasets DEGs intersection venn diagram,blue represents two data sets of GEO;yellow represents data sets of TCGA; D.Volcano plot of DEGs; red represents up-regulated genes;green represents down-regulated genes; abscissa represents fold change;ordinate represents P-value.

图3 GO及KEGG通路富集分析Fig.3 GO and KEGG pathway enrichment analysisNote:A.GO biological function enrichment analysis;redder represents smaller P-value,bluer represents larger P-value; abscissa represents number of genes,ordinate represents BP,CC,and MF;B.KEGG pathway enrichment analysis;color of dots represents fold change,size of dots represents number of genes,and connecting lines of different colors represent enriched pathways.

图4 GSEA通路富集分析Fig.4 GSEA pathway enrichment analysisNote:A.Pathway ridge map for GSEA enrichment analysis,color represents P-value,mountain shape represents distribution; B.Enrichment results of cell cycle and HIF-1 signaling pathway.

图5 PPI网络和hub基因Fig.5 PPI network and hub geneNote:A.PPI network analysis graph,node size represents clustering coefficient,larger node size represents larger clustering coefficient,greater proportion of genes in network represents greater node color representation,greater degree represents more connections node will have,greater blue representation,middle yellow,smallest orange; thicker connection represents higher score;thicker line represents more interaction between two proteins;B.Schematic diagram of hub gene,red indicates a high enrichment score,connection lines represent interactions between genes.

图6 10个hub基因的诊断价值分析Fig.6 Analysis of diagnostic value of 10 hub genesNote:A.ROC curves for AURKA,NCAPG,CDC20,CENPF and CCNB1;B.ROC curves for TPX2,CDCA8,DLGAP5,UBE2C and KIF23;Abscissa represents specificity and ordinate represents sensitivity.
2.3PPI网络构建及hub基因选择 STRING构建的PPI网络如图5A所示,利用cytoHubba插件选择的10个hub基因分别为 CCNB1、DLGAP5、CDC20、CENPF、KIF23、NCAPG、AURKA、CDCA8、UBE2C和TPX2(图5B)。
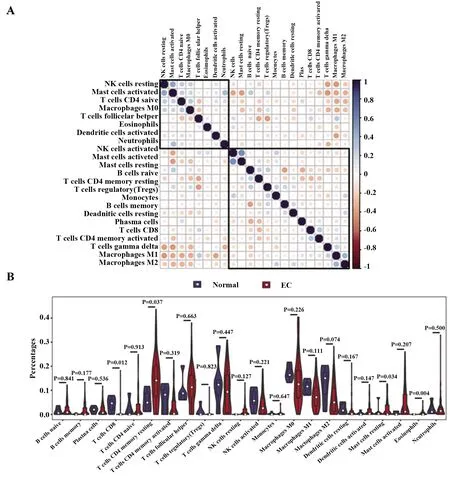
图7 免疫细胞浸润的相关性热图和小提琴图Fig.7 Correlation heat map and violin map of immune cell infiltrationNote:A.Correlation heat map of 22 kinds of immune cell infiltration; blue indicates positive correlation,red indicates negative correlation,darker color represents stronger correlation; both abscissa and ordinate represent 22 types of immune cells;B.A violin map of 22 immune cell infiltration ratios; red for EC sample group,blue for normal endometrial sample group,red marks indicate a difference in infiltration between two sets of samples; horizontal axis represents immune cells,vertical axis represents percentage of immune cell infiltration.
2.4诊断生物标志物的筛选 ROC分析结果显示,AURKA(AUC=0.804)、NCAPG(AUC=0.854)、CDC20(AUC=0.826)、CENPF(AUC=0.806)和 UBE2C(AUC=0.817)具有较高诊断价值(图6)。
2.5免疫细胞浸润结果 22种免疫细胞的相关性热图显示,M1型巨噬细胞与M2型巨噬细胞呈显著正相关,活化的肥大细胞与中性粒细胞呈显著正相关;M1型巨噬细胞与静止期NK细胞和活化的肥大细胞呈负相关,活化的肥大细胞与活化的NK细胞呈负相关,滤泡辅助性T细胞与静息期CD4+记忆性T细胞和调节性T细胞呈显著负相关(图7A)。22种免疫细胞浸润差异的小提琴图显示,与正常子宫内膜样本相比,静息期CD4+记忆性T细胞浸润较多,而CD8+T细胞浸润较少(图7B)。
3 讨论
EC是常见的妇科恶性肿瘤,发病率和死亡率较高[1]。EC的高死亡率在很大程度上归因于诊断不及时,因此寻找特异性早期诊断生物标志物对EC患者的预后改善至关重要。多个研究表明EC患者部分失调的基因可作为其诊断的生物标志物[35-37]。因此,本研究利用生物信息学工具对EC的mRNA表达谱进行分析,筛选出EC的诊断生物标志物。另外,越来越多的证据表明,多种免疫细胞与EC的发生和进展密切相关[16,38]。本研究采用CIBERSORT对EC进行免疫细胞浸润分析,初步探讨免疫细胞浸润在EC中的作用。
本研究从GEO和TCGA数据库下载EC的表达谱数据集,采用生物信息学方法共鉴定出448个DEGs。GO功能富集分析显示,DEGs主要涉及染色体分离、核分裂等生物学过程,介导趋化因子活性、丝氨酸肽酶活性等分子功能,DEGs基因产物主要富集于着丝粒、细胞间桥和细胞外基质。富集的通路主要与“Wnt信号通路”“TGF-β信号通路”“细胞周期信号通路”及“HIF-1信号通路”相关。Zhou等[39]研究表明SOX17通过Wnt信号通路抑制EC转移。多个研究表明,TGF-β信号通路在EC中被激活[40,41]。细胞周期信号通路激活可促进EC进展[42,43]。HIF-1信号通路是细胞在低氧环境中存活的关键通路,Seeber等[44]研究提示HIF-1信号通路与EC的临床预后不良有关,可作为EC的治疗靶点。以上研究结果与本研究结果相符,提示本研究结果准确性较高。
此外,本研究发现E2F1、CDC27和BUB1是细胞周期各通路中的重要参与者,VEGFA、AKT1和PIK3R2在HIF-1信号转导过程中起重要作用。E2F1是E2F转录因子家族成员,可调控细胞周期进程[45]。Song等[46]发现E2F1主要参与细胞周期调控,并可能促进EC的发生发展。后期促进的复合物/环体(APC/C)是有丝分裂期间蛋白质降解的主要调节剂,CDC27是APC/C的核心亚基,通过结合CDH1和CDC20激活APC/C,进而识别并降解目标底物[47]。Qiu等[48]研究表明CDC27通过调节p21表达控制G1/S期转变,在结直肠癌的增殖中起关键作用,且可作为结直肠癌患者的独立预后因素。BUB1是一种丝氨酸/苏氨酸激酶蛋白,在有丝分裂过程中起关键作用[49]。研究认为BUB1可作为胃癌的独立预后标志物[50]。E2F1、CDC27和BUB1均参与细胞周期的调控过程,而细胞周期通路又与EC密切相关,因此E2F1、CDC27和BUB1可能通过调控细胞周期参与EC发生发展。HIF-1可调节VEGFA表达,而VEGFA表达下调可抑制EC患者血管生成[51,52]。研究已经证实AKT1、PIK3R2突变在EC的发生发展中具有重要作用[53,54]。本研究表明AKT1、PIK3R2是HIF-1通路的重要参与者,因此推测AKT1、PIK3R2可能通过调控HIF-1信号通路参与EC进展。
本研究构建了DEGs的PPI网络,并确定CCNB1、DLGAP5、CDC20、CENPF、KIF23、NCAPG、AURKA、CDCA8、UBE2C和TPX2为hub基因,其中AURKA、UBE2C和CDC20已被证明与EC密切相关,AURKA过表达与EC的分级和组织学类型密切相关,可作为EC的治疗靶标和临床生物标志物,雌激素促进UBE2C介导的肿瘤上皮间质转化参与EC的进展[55,56]。Huo等[57]研究证实CDC20可作为EC的诊断生物学标记物。以上结果与本研究结果一致。CCNB1、DLGAP5、CENPF、KIF23、NCAPG、CDCA8和TPX2与多种肿瘤的发生及进展密切相关,但与EC的关系尚未明确,值得深入研究[58-64]。
本研究对hub基因进行诊断价值评估发现5个基因(AURKA、NCAPG、CDC20、CENPF及UBE2C)具有较高的诊断价值,其中AURKA、UBE2C及CDC20已被证实与EC密切相关,可能成为EC的诊断生物学标志物[55-57]。研究表明,NCAPG在肝细胞癌中表达上调,可作为肝细胞癌潜在的临床生物学标志物及治疗靶标[65]。CENPF被公认为肝细胞癌的早期诊断生物学标志物[66]。由此推测NCAPG和CENPF可能参与EC的发生及进展,有望成为EC的潜在新型诊断生物学标志物。
通过对EC中的免疫细胞进行相关性分析可知,活化的肥大细胞与中性粒细胞、M1型巨噬细胞及活化的NK细胞密切相关,提示其可能存在相互作用。Hughes等[67]发现肥大细胞可以介导早期中性粒细胞募集,表现出抗炎特性。另有研究表明,肥大细胞对白色念珠菌感染有反应且可调节感染期间巨噬细胞的吞噬作用[68]。登革热感染期间肥大细胞的免疫监视可促进NK细胞募集及病毒清除[69]。上述研究结果与本研究结果相符,但仍需进一步实验验证。
本研究通过免疫细胞浸润分析发现CD4+记忆性T细胞浸润增多和CD8+T细胞浸润减少可能与EC的发生发展有关。CD4+记忆性T细胞是治愈肿瘤的关键工具,其再次介导免疫应答可迅速产生大量CD4+T细胞,CD4+T细胞可直接杀死癌细胞,也可通过刺激和募集CD8+T细胞或其他免疫细胞间接杀伤癌细胞[70-72]。CD4+记忆性T细胞在机体免疫应答中由一部分初始T细胞分化而来,本研究发现EC中CD4+记忆性T细胞浸润增多,与上述观点一致。Ribatti等[73]研究发现EC中CD8+T细胞数量随着肿瘤进展而增加。然而,本研究分析结果显示EC中的CD8+T细胞数量减少,一方面可能由于CIBERSORT分析是基于有限的基因数据,这些数据可能会偏离细胞的异型性相互作用、疾病诱导失调或表型可塑性,另一方面其研究的EC样本仅有45例,检验效能偏低。因此,需要更大样本验证CD8+T细胞在EC中的浸润情况。
总之,本研究通过对EC的表达谱数据进行生物信息学分析,共获得448个DEGs和10个hub基因。进一步对hub基因进行ROC分析得到5个诊断生物学标志物(AURKA、NCAPG、CDC20、CENPF和UBE2C)。此外,免疫细胞浸润分析发现CD4+记忆性T细胞和CD8+T细胞可能参与EC的发生发展,为EC的新型诊断生物学标志物开发及进一步研究其分子机制提供了理论依据。作为未来的研究方向,课题组会在分子、细胞及组织水平上验证本研究结果的准确性,并进一步探讨5个诊断标志物与CD4+记忆性T细胞和CD8+T细胞的调控关系。
猜你喜欢
中国现代医生(2022年21期)2022-08-22
浙江医学(2022年8期)2022-05-21
健康体检与管理(2022年2期)2022-04-15
皮肤性病诊疗学杂志(2021年5期)2021-11-27
天津医科大学学报(2021年2期)2021-03-29
三农资讯半月报(2021年1期)2021-01-27
微创医学(2020年6期)2020-03-09
新课程·中旬(2018年2期)2018-04-19
CHINESE JOURNAL OF AERONAUTICS(2016年3期)2016-11-23
医学研究杂志(2015年7期)2015-06-22
