
嵌合抗原受体T细胞免疫疗法在肿瘤治疗中的研究进展①
2020-12-24 07:09张宸瑜陈柯远贾雨婷季少平
中国免疫学杂志 2020年22期
张宸瑜 陈柯远 贾雨婷 季少平
(河南大学基础医学院,开封 475000)
近年来,以T细胞为效应细胞的靶向性免疫细胞疗法成为抗肿瘤研究的热点,其目的是产生或者增强针对癌细胞的免疫应答来对抗癌症。这些技术也可称为过继性细胞转移(adoptive cell transfer,ACT)疗法。近几年来,此方法广泛被认为是最有可能征服肿瘤的免疫治疗技术。它主要是从人肿瘤内部或外周血中采取自身T淋巴细胞,进行基因工程修饰、加入细胞因子等刺激、进行体外培养,使具有一定抗肿瘤活性的T淋巴细胞大量扩增,再回输至患者体内,从而达到抗肿瘤效果。一般用于过继性免疫疗法的免疫细胞包括:淋巴因子激活的杀伤细胞(lymphokine-activated killer cell,LAK),细胞因子诱导的杀伤细胞(cytokine induced killer,CIK),细胞毒性T淋巴细胞(cytotoxic lymphocyte,CTL),肿瘤浸润性淋巴细胞(tumor infiltrating lymphocytes,TILs),T细胞受体(T cell receptor,TCR)T细胞和嵌合抗原受体(chemical antigen receptor,CAR)T细胞等。目前,TIL、TCR-T、CAR-T细胞技术在临床试验中已经取得了良好成效。
1 三种ACT疗法技术原理
1.1TILs细胞技术原理 TILs抗肿瘤技术是用机械处理或酶消化的方式,从肿瘤组织内部分离出TILs,在体外大量扩增后再回输至患者体内进行免疫治疗的方法。TILs是淋巴细胞的异质群体,主要由T淋巴细胞和自然杀伤细胞(natural killer cell,NK)组成,它可以自发迁移到任何实体肿瘤内部。据报道,使用自体肿瘤浸润淋巴细胞(TIL)的过继细胞转移技术在治疗黑色素瘤转移的患者时,能够达到约50%的反应率[1]。但最近有研究发现,在肠癌和卵巢癌分离出的TIL中,只有大约10%的TIL细胞有识别周围癌细胞的能力,其他的都是与癌细胞无关的T细胞。更意外的是,有两名患者的肿瘤组织样本中根本没有能识别癌细胞的杀伤性T细胞,尽管这两份组织样本中也充满了TIL[2]。由此可以得知,通过体外扩增后回输的TIL 细胞绝大部分在体内往往不能有效识别肿瘤细胞,因此TIL过继细胞转移技术仍需要进一步的研究与改善。
1.2TCR-T细胞技术原理 TCR-T细胞技术是通过转染能识别肿瘤相关抗原(Tumor-associated antigen,TAA)的TCR基因到宿主T细胞中,使T细胞表达能特异性识别TAA的TCR,并具有分泌细胞因子和特异性杀伤肿瘤细胞的能力。TCR是由α链和β链与T细胞表面的CD3复合物以非共价结合构成(图1)。在T细胞杀伤靶细胞的过程中需要抗原提呈细胞(antigen presenting cell,APC)递呈抗原,并形成抗原肽-MHC分子复合物,才能被TCR的可变区识别并初步活化T细胞;同时与T细胞接触的APC也被活化,上调表达共刺激分子CD80/CD86,与T细胞表面共刺激分子CD28相互作用,产生T细胞活化第二信号。此时T细胞被完全活化,开始诱导毒性T淋巴细胞分泌杀伤因子并有效杀伤靶细胞,或通过Fas/FasL途径诱导靶细胞凋亡(图2)。
TCR-T淋巴细胞的优点是可以识别所有能被人源APC呈递的HLA-Ⅰ类、Ⅱ类抗原,包括细胞内和细胞膜上的肿瘤相关抗原,更容易找到特异性高的“安全靶点”,副作用较小,也更容易侵入肿瘤内部。不过在有关TCR-T的研究中,其缺点也很明显——Johnson等[3]开发了针对MART-1表面抗原位点的高亲和力TCR, 可以较强识别低表达 MART-1的恶性黑色素瘤细胞。由于MART-1抗原的低特异性,虽然有一些疗效,但有超过一半的患者表现出皮肤、眼睛和耳朵中正常黑色素细胞破坏的症状,有时需要局部应用类固醇给药来治疗葡萄膜炎和听力丧失。

图1 TCR的结构示意图Fig.1 Schematic of TCR
1.3CAR-T细胞技术原理 CAR-T细胞免疫治疗技术是通过将患者的T淋巴细胞转入嵌合的CAR序列,表达后在体外大量扩增,然后回输入患者体内并杀死肿瘤细胞的技术。转入CAR序列,使得T细胞具有特异性识别肿瘤的能力。与TCR-T不同的是,CAR-T细胞能以非MHC限制的方式识别靶细胞,并激活自身增殖,分泌大量细胞因子,同时诱导靶细胞凋亡。
通常,CAR是由胞外用来识别肿瘤抗原位点的抗体可变区以及胞内来自T细胞的信号转导模块组合而成的融合蛋白。胞外的单链抗体轻、重链可变区scFv片段负责识别并结合肿瘤抗原靶点,跨膜区的铰链提供抗体识别的灵活性,胞内信号转导区的CD28和CD3ζ负责向细胞内传递活化信号和增殖信号。CAR-T细胞被激活后会在短时间内分泌大量细胞因子激活自身以及其他免疫细胞,并诱导靶细胞凋亡。
据报道,CAR已经发展到第四代(图3),第一代的CAR是1989年Eshha研究小组发明的,其胞外是scFv单链抗体片段,胞内只有CD3ζ。第一代CAR在细胞扩增、体内存活时间以及细胞因子分泌等方面存在一定缺陷,导致其临床疗效未达到预期效果;第二代CAR在第一代的基础上引入了共刺激分子胞内结构域[(costimulatory molecules endodom-ain)CD28或CD137(4-1BB)],与第一代相比,第二代CAR-T细胞比第一代具有更强的增殖能力、更高的细胞毒性以及更长的体内存活时间;第三代CAR是在加入CD28的同时,又加入了CD137或CD134,进一步延长体内T细胞的存活时间,增强其抗肿瘤能力。目前,第二代CAR-T技术是临床试验研究最常用的细胞免疫技术[4]。第四代CAR-T是在第三代CAR-T的基础上,同时导入可诱导表达的IL-12基因,使其获得CAR-T细胞与诱导型IL-12的双重抗癌活性。IL-12能以自分泌方式增强CAR-T细胞活化,还能以旁分泌方式吸引并激活先天性免疫细胞,而先天性免疫细胞则可以分泌TNF-α来攻击那些不被CAR-T细胞识别的阴性抗原肿瘤细胞,从而进一步加强了CAR-T细胞抗肿瘤的免疫能力[5]。
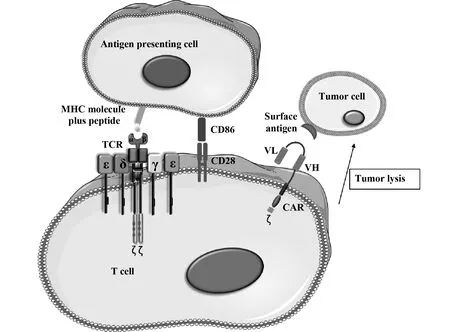
图2 T细胞活化的双信号途径与CAR-T细胞对肿瘤细胞的杀伤Fig.2 Double signal pathway of T cell activation and kill of tumor cells by CAR-T cells
目前,CAR-T细胞免疫技术已经在血液肿瘤方面取得了巨大成功,尤其是以CD19为靶点的CAR-T免疫疗法已经成功治愈慢性淋巴细胞白血病(Chronic lymphocytic leukemia,CLL)和急性淋巴细胞白血病(Acute lymphocytic leukemia,ALL)等血液恶性肿瘤。由于CD19主要表达于B淋巴细胞谱系起源的细胞,因此CD19-CAR-T在治疗效果和低毒性方面都具有优势。此外,针对实体瘤的CAR-T细胞疗法也具有一定成效,但是依然存在一些潜在障碍,其中包括:①CAR-T细胞不能特异的定位到肿瘤部位;②体内具有阻止CAR-T细胞浸润的物理屏障;③由于实体瘤抗原的高度异质性或缺乏特异性导致抗原选择难度增加;④一些健康器官/细胞中靶点抗原表达导致脱靶毒性的风险增高;⑤在肿瘤微环境中一些免疫抑制因子会使CAR-T细胞功能失调[6]。目前,正在进行的临床前研究和临床试验试图通过使用改良的基因转移方法和治疗方案来克服这些障碍。 开发新的更具有特异性的靶标进行 CAR

图3 四代CAR的结构示意图Fig.3 Schematic of four-generation CAR
的改造:例如用于结肠直肠癌的CEA、用于神经母细胞瘤和肉瘤的二唾液酸神经节苷脂GD2、用于前列腺癌和黑色素瘤的PSMA以及用于胶质母细胞瘤的EGFRvⅢ等,可以不同程度地提高抗原的特异性,降低毒副作用,有望增加治疗效果。
2 CAR-T细胞疗法的临床应用
2.1CD19-CAR-T 2011年起,靶向CD19的第二代CAR-T细胞技术成为CAR-T细胞免疫疗法的主流。CD19在B细胞谱系起源的恶性肿瘤中表达水平较高,并且它不在B细胞谱系之外的细胞表达,因此在一定程度上降低了脱靶毒性,这使得CD19成为非常理想的肿瘤相关抗原靶点[7]。但由于正常的B淋巴细胞也会有CD19的表达,所以经过CD19-CAR-T技术成功治疗的患者通常具有深度B细胞发育不良的症状,需要通过静脉注射免疫球蛋白来弥补B细胞的损失[7,8]。幸运的是,在停用CD19-CAR-T治疗后,大部分患者通过造血使得B淋巴细胞得以恢复。
ALL患者Emily是第一位成功接受CAR-T细胞治疗的人,她于2012年加入了宾夕法尼亚大学的CAR-T-19临床试验,成为全球第一位接受试验性CAR-T细胞免疫治疗的儿童患者。该患者接受治疗后曾一度出现细胞因子风暴;幸运的是在接受了降低免疫反应的药物依那西普(一种肿瘤坏死因子阻断剂)和托珠单抗(一种人源化白细胞介素6受体拮抗剂)后,CAR-T的严重副作用几乎被完全缓解,最终检查结果显示患者体内的癌细胞彻底消失,并且5年没有复发[9]。
2.2HER2-CAR-T 人表皮生长因子受体2(human epidermal growth factor receptor 2,HER2)作为乳腺癌靶点研究的比较多,但在骨肉瘤、尤文氏肉瘤等其他肿瘤治疗中也有一些研究。值得注意的是Ahmed等[10]研究发现,HER2低水平表达的恶性肿瘤也可以用HER2-CAR-T细胞进行治疗并且在临床前动物模型中这些HER2-CAR-T细胞具有有效的抗肿瘤活性[11]。在临床试验中,19名HER2阳性肿瘤患者输注HER2-CAR-T细胞后,普遍耐受性良好,HER2-CAR-T细胞可以在患者体内持续存在6周而没有明显的毒性,患者中位总生存期达到10.3个月(范围5.1至29.1个月)。这项研究首次评估出HER2-CAR-T细胞在癌症患者的治疗中具有安全性和有效性,为今后HER2-CAR-T细胞免疫疗法的癌症治疗研究奠定了基础[12]。
2.3GD2-CAR-T 二唾液酸神经节苷脂(disialoganglioside,GD2)少量表达于人小脑以及外周神经细胞中,而在一些肿瘤中,比如神经母细胞瘤、黑色素瘤等也有较高表达。GD2是FDA批准的治疗性单克隆抗体的靶标,可以作为细胞免疫治疗的肿瘤相关抗原。在Louis等[13]的研究中,19例恶性程度较高的神经母细胞瘤患者经过GD2-CAR-T细胞的输注治疗后,输注缓解8例,剩下复发的11例当中,又有3例完全缓解。在经过5年的随访中,唯一观察到与治疗相关的不良症状的2名受试者出现低度发热和轻度至中度的局部疼痛;1名受试者出现不明原因的局部疼痛,但没有受试者出现与GD2单克隆抗体输注相关的神经性疼痛或功能障碍。在这些病例中还观察到CAR-T细胞在患者体内可以存活很长时间(最长时间长达4年),表明CAR-T细胞可能在患者体内具有持久性,并且这种持久性似乎可以预防肿瘤复发,与患者更长的存活率相关。
2.4CAⅨ-CAR-T 碳酸酐酶Ⅸ(Carbonic anhydr-ase Ⅸ,CAⅨ)在肾癌细胞中表达量较高。Lamers等[14]将CAⅨ-CAR-T用于治疗转移性肾细胞癌(metastatic renal cell carcinoma,mRCC)。结果8名患者均发生CTC 2~4级肝功能异常。其中4名患者因脱靶作用严重而停止治疗,其肝组织活检的结果显示,表达CAⅨ正常的胆管上皮中伴有T细胞和CAR-T细胞的浸润。为了预防CAR-T的脱靶毒性,研究者经过实验发现,2~5 mg G250的用药剂量(98~320 ng/ml的血液水平),抗体可以有效覆盖在低表达CAⅨ的正常肝细胞表面,保护正常肝组织免受CAR-T细胞的伤害,并且该抗体水平并不阻止CAR-T细胞杀伤肿瘤细胞。如果要完全阻断CAR-T杀伤mRCC肿瘤细胞,则抗体在血液中的水平需要增加到10 μg/ml以上。后来研究者用低剂量的CAⅨ单克隆抗体(mAb)G250对另外4名患者进行预处理,再进行CAⅨ-CAR-T细胞的输注治疗,结果显示并未有肝毒性和外周T淋巴细胞浸润持续增强的迹象。该报告显示实体瘤的脱靶毒性症状可以在治疗前通过注射靶点mAb改善。未来,这一技术有可能是CAR-T技术攻克实体瘤的关键。
3 不良反应
3.1细胞因子释放综合征(cytokine release syndrome,CRS) CRS是一种由T细胞活化引发的全身性炎症反应。输注CAR-T细胞后患者体内的T细胞、B细胞、NK细胞以及单核/巨噬细胞会释放大量的细胞因子,主要是IL-1、IL-6、TNF-α、IL-12、IFN-α、IFN-β、IFN-γ、MCP-1和IL-8等。这些细胞因子水平显著升高,从而出现高热、肌痛、乏力、恶心、低血压、心动过速、肾损害、肝衰竭,甚至呼吸困难等不良症状,严重者会有致死性风险。CRS通常发生在CAR-T治疗后的第一周内,且于1~2周内达到最高峰,其严重程度与肿瘤负荷相关。最近Theodoros等[15]发现CRS是由炎性分子IL-1引发的,在治疗方案中加入抑制IL-1的阿那白滞素(IL-1抑制剂,Anakinra)能够有效控制CRS和神经毒性。也有研究发现,细胞因子的释放与肾上腺素、去甲肾上腺素、多巴胺等儿茶酚胺类物质有关,通过限制此类物质的合成可以有效控制CRS的发生。在多种免疫治疗的小鼠模型中,这种方法都能够降低细胞因子产生、增加存活率,同时不会影响到治疗效果。并且,儿茶酚胺合成的关键限速酶酪氨酸羟化酶(TH)抑制剂α-甲基酪氨酸(MTR)以及α-肾上腺素受体阻滞剂哌唑嗪等物质是FDA批准用于治疗高血压的药物[16-18]。
3.2神经毒性征 CAR-T细胞疗法的另一严重副反应就是神经毒性。神经毒性在很大程度上是可逆的,在使用托珠单抗和地塞米松(一种糖皮质激素,主要用于过敏性与自身免疫性炎症性疾病)进行治疗后,神经毒性一般会于4.5 d(中位天数)后完全缓解。大部分患者的神经毒性并发症是短暂和轻微的,只有小部分患者会有严重副反应,包括昏迷、头痛、意识水平下降、失语症、语言障碍、神经错乱、癫痫、局灶性神经功能缺损和颅内大出血等。在Gust等[19]的研究中,CD19-CAR-T用于治疗133名患有难治性CD19阳性B细胞急性淋巴细胞白血病(B-ALL)、CLL和非霍奇金淋巴瘤(NHL)的患者,虽然在Ⅰ期研究中取得较高的反应率,但有40%的患者发生了不同程度的神经毒性,在这40%的患者当中又有90%的患者同时发生了CRS,并且更早出现CRS的患者会有更高的神经毒性风险。目前神经毒性的根本原因尚不清楚,不过研究发现,有内皮细胞活化的患者经过CD19-CAR-T治疗后可能会发生更为严重的神经毒性,且使用托珠单抗等药物治疗后恢复更慢,说明神经毒性与患者内皮活化存在一定联系[19]。
3.3脱靶毒性 有一些TAA位点,不仅在肿瘤细胞表面表达量较高,而且正常组织细胞表面也会有表达。因此CAR-T细胞在杀伤肿瘤细胞的同时也可能会损伤正常组织细胞,这就产生了脱靶毒性。碳酸酐酶Ⅸ(CAⅨ)在肾透明细胞癌表面高表达,但是正常胆管上皮细胞表面也会表达,Lamers等[14]在研究靶向CAⅨ的第一代CAR-T细胞的试验中,发现转移性肾细胞癌的患者经过治疗后,出现因T淋巴细胞浸润导致的胆管炎和一定程度的肝毒性。
同样在靶向HER2位点的第三代CAR-T细胞疗法试验中,也存在显著脱靶毒性。研究显示其毒性是由CAR-T细胞识别并杀死低密度表达的阳性HER2肺上皮细胞引起的,同时引发肺衰竭和大量细胞因子的释放。但是Ahmed等[12]同时也发现低亲和力的scFv配合低剂量的CAR-T细胞进行治疗是相对安全的,但是其临床抗肿瘤活性较低,这就迫切需要寻找特异性更强的肿瘤相关抗原位点,或者研究出有效的靶点单抗对患者进行预处理保护患者正常组织来降低脱靶毒性[14]。
4 癌症复发
在免疫治疗中,肿瘤逃逸是癌症复发的主要原因。虽然大多数复发性的白血病患者在经过CD19-CAR-T细胞治疗后可以完全缓解,但在一些ALL患者当中,CAR-T细胞靶向的CD19阳性肿瘤细胞上抗原表位的缺失会导致CAR-T细胞无法正常识别B-ALL肿瘤细胞,进而导致癌症复发,复发后的肿瘤细胞为CD19阴性。在Grupp[9]的研究当中,两名患有复发和难治性ALL的儿童经过CAR-T治疗后,一名患者持续缓解,另一名患者一年后复发,且复发患者的肿瘤细胞不再表达CD19。Jacoby等[20]通过对小鼠B-ALL肿瘤模型的研究表明,CD19-CAR-T的作用压力可诱导B-ALL肿瘤细胞发生部分或完全的免疫表型改变,引起CD19表达降低或者沉默,从而耐受CD19-CAR-T治疗。CD19的缺失表明对于ALL患者的治疗除了靶向CD19之外还需要靶向其他分子进行双靶点或多靶点CAR-T治疗。
近期研究发现在给一名身患复发/难治性B细胞急性淋巴细胞白血病(B-ALL)的患者构建CAR-T细胞时,CAR基因意外地被加到患者癌变的B细胞上,形成了“CAR-癌细胞”。并且加到癌细胞上的CAR会与癌细胞表面的CD19结合而屏蔽CD19分子,使得CAR-T细胞失去了识别癌细胞靶标的机会。虽然这名患者在接受CAR-T细胞回输的28 d之后,病情完全缓解,但在261 d之后由于“CAR-癌细胞”大量增殖,癌症复发。患者最终死于与白血病相关的并发症[21]。
5 通过基因编辑改良CAR
改变CAR的亲和力。通过改变CAR对肿瘤靶点的亲和力,能够使CAR-T细胞更容易区分健康组织细胞上的低密度抗原分子以及肿瘤细胞上面的高密度抗原分子。比如在Richman等[22]研究的GD2-CAR-T临床模型中,虽说GD2特异性CAR结合抗原的能力被增强,并且增加了抗肿瘤活性,然而,这种增强的体内抗肿瘤功效也伴随着致死性的中枢神经系统(central nervous system,CNS)毒性。其中包括脑内CAR-T细胞的大范围浸润和增殖以及对神经元的破坏。因此既能保证抗肿瘤能力又能控制好脱靶毒性的CAR-T细胞是最佳选择,这些还需要进一步的优化和探索。
消除免疫抑制信号,如CTLA-4和PD-1。CTLA-4(CD152)表达于活化的CD4+和CD8+T细胞,其配体与CD28一样都是CD80和CD86,但CTLA-4与配体结合的亲和力显著高于CD28,并传递抑制性信号,起下调或终止T细胞活化的作用;PD-1(programmed death 1)也表达于活化T细胞,配体为PD-L1和PD-L2,PD-L1与配体结合后可抑制T细胞的增殖分化及Ig的分泌。故可尝试利用基因编辑技术先将T细胞的这两段基因沉默,再转进CAR基因构建CAR-T,从而达到保持CAR-T细胞活性的目的[23]。
靶向两种或多种抗原位点。靶向单个抗原位点的CAR-T细胞在区分肿瘤细胞和健康组织细胞的能力方面会有一些限制。为了增强针对肿瘤细胞的特异性,可以开发靶向两种或多种抗原位点的联合识别技术。比如对B-ALL患者的治疗过程中,可以使用CD19+CD22双靶点CAR-T疗法,即使肿瘤B细胞上的CD19抗原表位缺失,还可以通过靶向CD22来避免肿瘤逃逸[24]。如果产生脱靶效应,则可以用mAb封闭靶点抗原来缓解脱靶效应。最早的双靶点CAR-T技术是将二代CAR的共刺激信号CD28与CD3ζ分开,即1个CAR-T细胞表达两种CAR。CAR1的scFv识别靶点1,其胞内区是CD3ζ,就像第一代CAR;CAR2的scFv识别靶点2,其胞内区只有CD28或CD137。当肿瘤细胞上的两种靶点被双靶点CAR-T细胞同时识别时,CAR-T细胞才可以完全活化并且杀伤肿瘤细胞[25]。
给CAR-T细胞添加自杀基因。Stasi等[26]设计了一种诱导型T细胞安全开关,它是基于人Caspase 9与修饰过的人FK结合蛋白的融合,可以条件性二聚化。当CAR-T细胞暴露于合成的二聚化药物时,诱导型半胱天冬酶9(iCasp9)被激活并诱导表达该基因的细胞快速死亡。研究显示在过继性细胞转移疗法中,一旦发生移植物抗宿主病,患者只要接受一种生物惰性小分子二聚化药物便可以在30 min内消除体内90%的转基因T细胞。因此通过iCasp9细胞自杀系统能够提高细胞免疫疗法的安全性,可以考虑将其拓展到临床应用上。
6 通用型CAR-T细胞
主要组织相容性复合体(major histocompati-bility complex,MHC)是一组决定移植组织是否相容的基因群,人的MHC称为人类白细胞抗原(human leukocyte antigen,HLA)基因复合体。由于MHC固有的限制性,ACT的治疗策略只能体现在自体T细胞,如果可以消除MHC的限制,那么可以使用健康供体的T细胞来构建通用型CAR-T。由健康供体T细胞构建的CAR-T具有许多克服与癌症治疗相关的免疫缺陷的潜力,相对患者自体T细胞具有显著的优势。同时能够简化CAR-T的制造工艺,有可能出现即用型的“CAR-T产品”,使得整个治疗过程更快捷,更便宜[27]。
根据Torikai等[28]的研究报道,有两名患者成功使用了通用型CD19-CAR-T细胞,但由于患者自身的免疫反应会使得自身NK细胞去裂解输注的CAR-T细胞,所以输注的CAR-T细胞数量会受到限制,因此治疗效果也很有限。为了阻止CAR-T细胞被患者的NK细胞裂解,我们可以利用基因编辑技术将HLA-E基因插入CAR-T细胞,再删除CAR-T细胞的HLA-Ⅰ类基因(HLA-A、HLA-B、HLA-C)。HLA-E分子是NK细胞表面CD94/NKG2的专一型配体,可以抑制NK细胞对表达HLA-E分子的细胞的杀伤作用[29]。HLA-Ⅰ类基因表达于所有有核细胞,可以被宿主CD8+T淋巴细胞特异性识别并杀伤。因此将HLA-Ⅰ类基因删除则可以避免CAR-T细胞被接受者的自身CTL识别和裂解,降低抗肿瘤活性。所以理论上这两种途径可以防止接受者自身T细胞杀死通用型CAR-T细胞。
7 结语
近期CD19特异性CAR-T细胞疗法已经被美国食品和药物管理局(FDA)批准用于治疗难治性B细胞前体ALL和弥漫性大B细胞淋巴瘤,也是第一种获得美国FDA商业批准的基因治疗方法。考虑到细胞因子综合征和神经毒性的风险,FDA要求医生完成CAR-T不良反应的救护培训,进而在CAR-T细胞的治疗过程中降低风险。
今后CAR-T的发展有可能与一些新型免疫治疗方法相结合,比如免疫检查点抑制剂等;发展即用型CAR-T细胞;也有可能将CAR-T细胞作为药物载体来靶向输送抗癌药物/蛋白,从而达到精准治疗的目的[30]。开发CAR-T最大的挑战就是缺乏临床前模型来评估CAR-T的安全性和有效性,尽管CAR-T细胞在血液肿瘤方面的研究比较成功,但用于实体瘤方面还是存在一定的障碍。随着科学家对肿瘤特异性抗原研究的不断深入以及基因修饰技术的不断发展,未来的CAR-T细胞免疫疗法将会更加精准有效。
猜你喜欢
当代水产(2022年6期)2022-06-29
中老年保健(2021年3期)2021-12-03
当代水产(2021年6期)2021-08-13
昆明医科大学学报(2021年2期)2021-03-29
昆明医科大学学报(2021年1期)2021-02-07
中国生殖健康(2020年7期)2020-12-10
中国兽医杂志(2019年5期)2019-09-18
小哥白尼(野生动物)(2019年5期)2019-08-27
生物学教学(2018年10期)2018-11-29
医学研究杂志(2015年7期)2015-06-22
