
Genetic dissection of husk number and length across multiple environments and fine-mapping of a major-effect QTL for husk number in maize(Zea mays L.)
2020-12-22 05:24:02GungfeiZhouYuxingMoLinXueGuoqingChenHuhuLuMinglingShiZhenlingZhngXiolnHungXudongSongDerongHo
The Crop Journal 2020年6期
Gungfei Zhou, Yuxing Mo, Lin Xue,b, Guoqing Chen,b, Huhu Lu, Mingling Shi,Zhenling Zhng, Xioln Hung, Xudong Song, Derong Ho
aJiangsu Yanjiang Institute of Agricultural Sciences, Nantong 226541, Jiangsu, China
bJiangsu Collaborative Innovation Center for Modern Crop Production, Nanjing 210095, Jiangsu, China
Keywords:Zea mays L.Husk number Husk length Quantitative trait locus Fine mapping
A B S T R A C T Husk number (HN) and husk length (HL) influence the mechanical harvesting of maize grain. We investigated the genetic basis of HN and HL using a population of 204 recombinant inbred lines phenotypically evaluated in five environments. The two husk traits showed broad phenotypic variation and high heritability. Nine stable quantitative trait loci(QTL)were identified by single-environment mapping,comprising four QTL for HN and five for HL, and three QTL explained >10% of the phenotypic variation. Joint mapping revealed 22 additive QTL and 46 epistatic QTL. Both additive and epistatic (additive ×additive) effects as well as a few large-effect QTL and some minor-effect QTL appeared to contribute to the genetic architecture of HN and HL. The QTL for HN located on chromosome 7, qHN7, which accounted for ~20% of phenotypic variation, was detected in all five environments. qHN7 was fine-mapped to a 721.1 kb physical region based on the maize B73 RefGen_v3 genome assembly. Within this interval, four genes associated with plant growth and development were selected as candidate genes.The results will be useful for improvement of maize husk traits by molecular breeding and provide a basis for the cloning of qHN7.
1.Introduction
Maize (Zea mays L.) is a forage crop, an energy crop, and an industrial raw material grown worldwide [1]. Agronomic traits, such as morphological traits [2], maturity traits [3],and yield component traits [4], have historically been the primary focus of geneticists and breeders of maize.Recently,Chinese maize breeders have increasingly concentrated on the husk,as it is a vital trait determining whether a variety is suitable for mechanical harvesting of the grain[5–10].Several husk traits, including husk number (HN), husk length (HL),and husk weight (HW), have been reported to be associated with kernel moisture content at harvest stage [8,9]. Proper shedding of husks is beneficial to kernel dehydration after physiological maturity and has no effect on kernel weight[10].
Researchers have focused more on the physiological function of the husk than on its application in maize breeding[11–18]. As an integral part of the ear, the husk is the most effective photosynthetic organ of maize,having higher carbon assimilation efficiency and a greater contribution to kernel dry matter, per unit area, than do leaves [11–13]. Suitable tightness and coverage of the husk protects the ear from invasion by diseases and insects[14,15].In addition,the husk maintains a suitable temperature for the growth and development of the ear[16],and directly or indirectly supplies fiber and anthocyanin for industrial and nutritional production[17,18].
Although the husk tightly encloses the ear, to date few studies have focused on detection of genetic loci underlying husk traits, in comparison with ear traits. Zhou et al. [6]identified eight and nine stable single-nucleotide polymorphisms (SNP) for husk number and weight, respectively, in a genome-wide association study (GWAS). Similarly, Cui et al.[19] identified nine SNP significantly associated with four husk traits using GWAS.Cui et al.[20]predicted five candidate genes for husk traits by combining linkage analysis and GWAS.
The husk, which refers to the leaves that extend from modified leaf sheaths and enclose the ear [11], develops from the lateral meristem and differs from foliar leaves,which are initiated from the shoot apical meristem[21].The husk architecture comprises several traits including HN,HL,HW, and husk tightness (HT). These husk traits are affected by initiation and elongation of the lateral meristem, involving cell division, differentiation, and metabolism. Thus,numerous quantitative trait loci (QTL) and genes are implicated in regulating the development of husk architectural traits [19].
QTL mapping has been successfully applied to detect genetic loci underlying complex phenotypic traits in many crops [22]. In combination with positional cloning, QTL mapping is an efficient strategy for identifying underlying genes[23–25].Together with advances in maize chip technology and reduction of testing costs, SNP markers have been extensively adopted owing to their high density on chromosomes,stable inheritance,and utility for automatic detection.Combining co-segregating SNP markers into adjacent bins based on single recombination events [26], enables such bins to be used as a markers for constructing a high-density genetic map. The bin-map method is more powerful for identification of QTL than conventional methods and has been employed for mapping QTL of diverse traits in maize,such as disease resistance[27],kernel traits[28,29],and plant architecture-related traits[2,30,31].
The objectives of the present study were to(1)estimate the genetic variance and heritability of HN and HL in a recombinant inbred line (RIL) population, (2) dissect the genetic architecture of HN and HL, (3) fine-map a major-effect QTL for HN,and(4)predict candidate genes involved in HN.
2. Materials and methods
2.1. Plant materials and field experiments
A RIL population composed of 204 lines, developed from a cross between the maize inbred lines DH4866 and T877,was used as the QTL mapping population. DH4866, the female parent of the Chinese elite hybrid Denghai 1, was derived from a cross between the two Chinese inbred lines 7922 and Ye478. T877 is an inbred line derived from a cross between the U.S. hybrid 78,599 and the Chinese inbred line E28 [8].
The RIL population and its parents were planted in five environments in China: Xuzhou, Jiangsu province (34°N,117°E) in 2015, Nantong, Jiangsu province (31°N, 120°E) in 2016 and 2017, and Sanya, Hainan province (18°N, 108°E,) in 2016 and 2017, hereafter referred to as 15XZ, 16NT, 17NT,16SY, and 17SY. Each line was grown in single rows 3.0 m in length with 0.6 m between rows at a planting density of 65,000 plants ha−1, following a randomized complete block design with two replications per environment. The HN and HL of 10–12 plants of uniform growth in the center of the rows were measured at the harvest stage.The HN was counted from the outermost to the innermost layers of the husk. The HL was measured for the longest layer of the husk from the tip to the base. Agronomic management of the field experiments was identical in each environment.
YD145,an inbred line derived from the RIL population that carried the T877 allele of qHN7 and the DH4866 allele for three additional QTL for HN,was selected to backcross with DH4866(as the recurrent parent)to develop a BC1F2population.A total of 2566 BC1F2plants were grown at Sanya in 2018 for finemapping of the physical location of qHN7.
Seeds of 54 homozygous recombinants selected from 2566 BC1F2plants were planted at Nantong in 2019 for progeny testing. The planting scheme and experimental design followed those used for the RIL population.
2.2. Phenotypic data analysis
The statistical analysis was performed using R 3.1.1 (https://www.r-project.org/). Analysis of variance (ANOVA)of HN and HL was performed using the lmer function of the lme4 package of R based on the following model: yij= μ + gi+ ej+ geij+ εij,where yijis the trait measured, μ is the grand mean over all environments,giis the genotypic effect of the ith genotype,ejis the location effect of the jth location, geijis the genotype ×enviroment interaction effect,and εijis the residual error.The broad-sense heritability (H2) of the two traits were calculated using the following formula: H2(%) = σ2g/(σ2g+ σ2ge/n + σ2e/nr) × 100%, where σ2gis the genotypic variance, σ2geis the variance of the interaction of genotype with location,σ2eis the error variance, n is the number of locations, and r is the number of replications [32]. To minimize environmental effects, best linear unbiased prediction (BLUP) values for HN and HL of each line across all environments were estimated with the same ANOVA model. All analyses, including phenotypic data, correlation, and ANOVA, were based on BLUP values.
2.3. Genotyping, bin map construction, and molecular marker development
The RIL population and two parental inbred lines were genotyped using an Affymetrix microarray, the CGMB56K SNP Array, which contains 56,000 maize SNPs, by China Golden Marker (Beijing) Biotech Co., Beijing, China. After quality control, 9780 SNPs were polymorphic between DH4866 and T877.Co-segregating SNP markers were assigned to single recombination bins using a custom Perl script.A total of 1868 recombination bins were assigned in the RIL population.They were used to construct a genetic linkage map,using the Kosambi mapping function to calculate the genetic distance between markers. Details of the construction of the bin map have been reported previously[33].
For fine-mapping of qHN7, based on the maize B73 RefGen_v3 genome assembly and resequencing information for DH4866 and T877, six polymorphic markers, comprising one simple sequence repeat (SSR) and five insertion/deletion(InDel) markers, were developed to identify the genotypes of the BC1F2plants using Primer 3 web version 4.1.0 (http://bioinfo.ut.ee/primer3-0.4.0/)(Table S1).
2.4. QTL analysis
Mapping of QTL in single environment was conducted using composite interval mapping (CIM) as implemented in Windows QTL Cartographer 2.5 [34]. The entire genome was scanned every 0.5 cM with a window size of 10 cM. Model 6 of the Zmapqtl module was selected to detect QTL and their effects. Forward–backward stepwise regression with five controlling markers was used to control the background from flanking markers. The confidence interval of QTL positions was estimated with the 1.5-LOD support interval method. To detect additive effects (A), additive × additive epistatic effects (AA) and QTL × environment interact effects(QEI), joint mapping analysis under multiple environments and epistasis analysis were performed using respectively ICIM-ADD and ICIM-EPI of the MET functional module in QTL IciMapping v4.1 software [35]. The threshold logarithm of odds(LOD)value was determined empirically at a significance level of P < 0.05 by 1000 permutations in all analyses.
QTL for a target trait identified in multiple environments with clearly similar positions(in overlapping 1.5-LOD confidence intervals provided by software)were considered the same[36].
2.5. Candidate gene analysis
Based on B73 RefGen_v3 in MaizeGDB(http://www.maizegdb.org),the genes within the refined the largest-effect QTL region were extracted and annotated by NCBI(https://www.ncbi.nlm.nih.gov).
3. Results
3.1. Phenotypic variation and heritability of HN and HL
Descriptive statistics for the HN and HL in the RIL population are presented in Tables 1 and S2. Significant differences in HN and HL were observed between the two parents (Table S2). Based on BLUP values, compared with those of DH4866 (HN was 10.64 and HL was 30.27 cm), in T877(HN was 6.72 and HL was 20.85 cm) HN was ~37% lower and HL was ~31% shorter. Broad phenotypic variation was observed in the RIL population, ranging from 5.91 to 10.91 in HN and from 18.93 to 31.73 cm in HL.
The variances of genotype (σ2g) and genotype × environment (σ2ge) were significant at P < 0.01 for HN and HL, and the broad-sense heritability of each trait was high (Table 1).
HN and HL approximately fitted normal distributions with little skewness and kurtosis observed, except for skewness of HL in 16NT, suggesting that the two traits were controlled by multiple loci (Figs. 1 and S1, Tables 1 and S2). No significant correlation between HN and HL (r2= 0.07) was observed, but each trait was significantly correlated among the five environments (Fig. S1).
3.2. QTL mapping of HN and HL in individual environment
Nine QTL associated with the two husk traits were identified at empirical LOD thresholds of 4.2 and 4.6 for HN and HL,respectively (Fig. 2, Table 2). The phenotypic variation explained by QTL ranged from 6.2% to 19.6%, summing to 43.0% and 46.5% for HN and HL, respectively.
For HN, four QTL, located on chromosomes 1, 3, 6, and 7,were identified. The QTL on chromosome 7, qHN7, showed the largest effect and explained 19.6% of phenotypic variation,with the DH4866 allele at this locus showing an additive effect of 0.67 increase in HN. For HL, five QTL, located on chromosomes 2, 3, 4, and 7, were identified. Two QTL on chromosome 4, qHL4.1 and qHL4.2, explained >10% of the phenotypic variation.
To further confirm the nine QTL, we also identified QTL that were significant in single environments (Fig. S2, Fig. S3,Table S3). Notably, qHN7 was detected in all five environments and explained 11.5%–20.3% of phenotypic variation.Two QTL, qHN1 and qHL3, were detected in three environments. Four QTL, qHN3, qHN6, qHL4.1, and qHL4.2, were detected in two environments. The remaining two QTL,qHL2 and qHL7, were detected in one environment. In addition to these nine QTL, two QTL for HN located on chromosomes 8 and 9 were identified in 17NT, and three QTL for HL on chromosomes 3, 4, and 8 were identified in 16SY,15XZ, and 16NT, respectively.
3.3. Joint mapping for HN and HL under multiple environments
A total of 22 additive QTL associated with the two husk traits were identified at LOD thresholds of 6.7 and 7.2 for HN and HL, respectively (Table S4). The phenotypic variation explained by each additive QTL ranged from 0.7% to 10.0%,with sums of 41.1% and 39.3% for HN and HL, respectively. The contributions of interaction between each additive QTL and environment (AE) ranged from 0.2% to 3.67%, with sums of 8.3% and 25.0% for HN and HL, respectively.
Nine additive QTL for HN were mapped on chromosomes 1,3, 5, 6, 7 and 9, including five identified in single-environment analyses. The phenotypic variance explained by each additive QTL was higher than those explained by AE.

Table 1–Descriptive statistics for analysis of variance and heritability of husk number and length in RIL population.
For HL, 13 additive QTL, located on chromosomes 1, 2, 3, 4,7, and 9, were identified. Seven QTL were localized to the same intervals as those identified by single-environment mapping. The phenotypic variance explained by each additive QTL was higher than those by AE except for one QTL.
3.4. Epistatic QTL-by-environment interactions for multiple environments
A total of 46 epistatic QTL associated with the two husk traits were identified at empirical LOD thresholds of 9.6 and 8.6 for HN and HL, respectively. The phenotypic variation explained by each epistatic QTL ranged from 1.0% to 4.4%,with sums of 43.8% and 54.1% for HN and HL, respectively. The contributions of interaction between each epistatic QTL and environment (AAE) ranged from 0.1% to 0.9%, with sums of 8.6% and 11.6% for HN and HL, respectively (Table S5).
For HN, 19 epistatic QTL were identified, involving 37 genetic loci. Fifteen QTL showed epistatic interaction across linkage groups, and the remaining four were in a single linkage group. The phenotypic variance explained by each epistatic QTL was higher than those explained by AAE.
For HL, 27 epistatic QTL were identified, involving 51 genetic loci. Twenty-four QTL showed epistatic interaction between different linkage groups, and the remaining three QTL were in a single linkage group. The phenotypic variance explained by each epistatic QTL was higher than those by AAE.
3.5. Fine mapping of qHN7
Based on QTL mapping in the RIL population, qHN7 was inferred to be a reliable and major-effect QTL for HN.Initially,qHN7 was mapped between the markers PZE-107047562 and SYN38586, which spanned a genetic distance of 21.4 cM,corresponding to a physical distance of 23.8 Mb in the B73 RefGen_v3 genome assembly (Fig. 3a). Fourteen polymorphic SNP markers [33] derived from a high-density linkage map and evenly distributed in this interval were selected and the 204 RIL lines were categorized into seven haplotypes, after removal of haplotypes consisting of fewer than three lines(Fig.3b).HN means of plants with and without DH4866 alleles were compared using BLUP values and a two-sample t-test.The HN values of H4,H5,H6,and H7 were significantly higher than that of H1, which did not carry DH4866 alleles at any of the 14 SNPs,whereas H2 and H3 showed similar HN values to that of H1 (Fig. 3c). This finding localized qHN7 to a 6.5 Mb interval flanked by PZE-107057279 and SYN37179 in the B73 RefGen_v3 genome assembly.
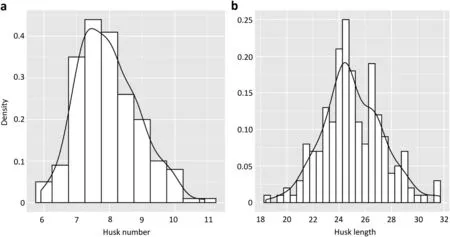
Fig. 1 – Frequency distribution of husk number and length in the RIL population.
To further restrict the target region of qHN7, a large BC1F2segregating population with 2566 plants was developed and genotyped using six newly developed markers (Table S1).Based on the genotyping results with the six markers in the interval containing qHN7,54 homozygous recombinants were divided into eight recombinant types (R1–R8) (Fig. 3d). The same analytical method, based on the phenotype of eight recombinant types in Sanya and Nantong, showed the HN values of R5,R6,R7,and R8 to be significantly higher than that of R1,which did not carry DH4866 alleles at any of the six new markers,whereas R2,R3,and R4 showed HN values similar to that of R1 (Fig. 3e). qHN7 was accordingly confined to the marker interval between I22 and I33 with a physical length of 721.1 kb in the B73 RefGen_v3 genome assembly.
3.6. Candidate genes in the qHN7 region
Based on the available annotation of B73 RefGen_v3,there are 32 predicted genes in the 721.1-kb target region(Table S6).Of these genes, 14 encode proteins of unknown function, 13 are transposable elements, and only five protein functions have so far been assigned.
4. Discussion
4.1.HN and HL are two important traits in maize mechanized breeding
The husk surrounding the ear of maize provides favorable conditions for the growth and development of the ear[14–16].Moisture evaporating from the kernels must pass through the husk at the dehydration stage [6]. Unfavorable husk traits,such as too many layers in maize cultivars limits the speed of dehydration of kernel after physiological maturity, resulting in a higher kernel moisture content at harvest stage. This condition impedes mechanical harvesting and represents the major barrier to maize development in China[5,7].

Table 2–Stably detected QTL for husk number and length in the RIL population.
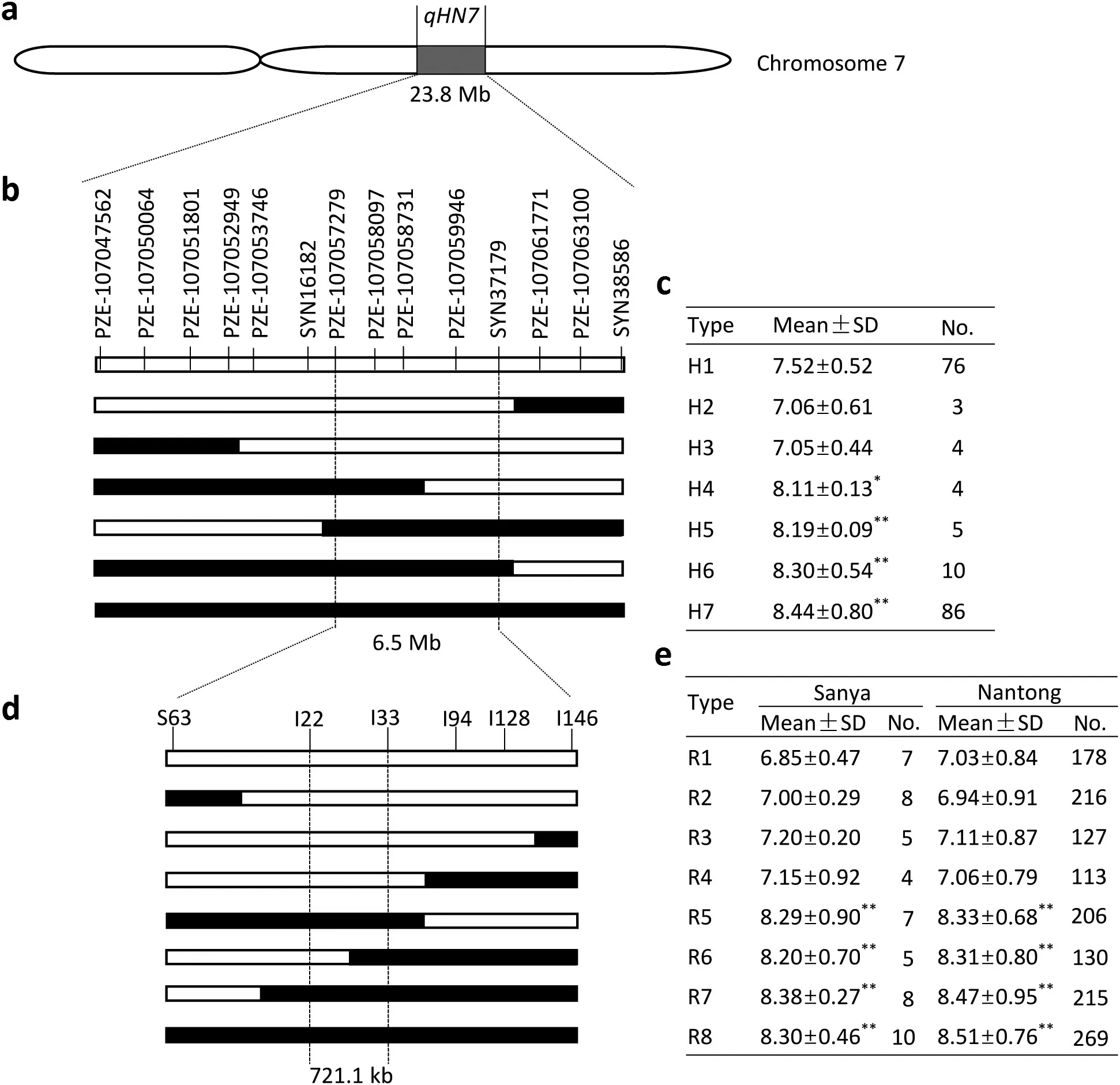
Fig.3–Fine mapping of qHN7.(a)qHN7 on maize chromosome 7.(b)Graphical genotypes of seven haplotypes.White segments represent genomic segments from T877 and black segments represent those from DH4866.Recombination breakpoints were placed midway between flanking markers.(c)Mean values of husk number for each haplotype.No.represents the number of lines and* and**represent statistical significance at P < 0.05 and P < 0.01,respectively. (d) Graphical genotypes of eight homozygous recombinants(see explanation for panel b)(e)Mean values of husk number for each homozygous recombinant in Sanya and Nantong,No.represents the number of plants and**represents statistical significance at P < 0.01.
Maize kernel moisture content at harvest stage showed significant positive correlations with HN and HL [6]. Appropriate reduction of HN and shortening of HL are conducive to loss of kernel moisture after physiological maturity without reducing kernel weight allowing maize kernels to be more easily threshed and less readily damaged during combine harvesting and to easier removal of the husk to ensure the quality of the seed [5–7,10]. These findings suggest that HN and HL are important traits for breeding maize for mechanized harvesting. Thus, dissecting the genetic basis of these two traits is beneficial for genetic improvement of maize for mechanical harvesting of kernels.
4.2. The RIL population was suited for QTL mapping of maize HN and HL
Development of an appropriate mapping population is the first critical step in QTL analysis. In this study, a maize RIL population with high recombination frequency [33] was used for the genetic dissection of two husk traits,HN and HL.Broad variation in the two traits was observed in the RIL population derived from two maize elite inbred lines, DH4866 and T877,which showed large differences in the two husk traits (Fig. 1,Table S1), and provided the necessary foundation for QTL mapping of HN and HL. A previous study [37] showed that relatively high heritability is crucial in determining the power of QTL detection. The high broad-sense heritability of HN and HL in the RIL population suggested that much of the phenotypic variation of these two traits was under genetic control.
4.3. Genetic complexity of maize HN and HL
The QTL identified showed inconsistent expression in different environments, a prominent characteristic of quantitative traits controlled by polygenes. Repeated detection of a locus in multiple environments is conventionally required for recognition of genuine and stable QTL [38,39]. The HN and HL of the RIL population were evaluated in five environments and nine QTL were detected. Only one (qHN7) was detected in all environments. This finding could be explained in at least two ways. One is QEI due to the specific QTL being expressed in one environment but not in another environment or strongly expressed in one environment and weakly expressed in another environment, a common property of many QTL,even for highly heritable traits [31,39]. However, compared to the genetic effects (A and AA), QEI, including AE and AAE,contributed only slightly to the phenotypic variance of HN and HL (Tables S4 and S5). The second explanation is rejection of positive loci with minor effect in different environments at a stringent detection threshold (4.2 for HN and 4.6 for HL). From Figs. S2 and S3, the remaining eight QTL still showed clear LOD peaks in different environments, although the LOD values of some QTL were lower than the threshold. Most of the QTL could also be detected by joint mapping in multiple environments (Table S4), and were accordingly considered stable. With the selected threshold of 2.5 for both traits, a common threshold in QTL mapping [36,40,41], all QTL could be identified in four or all five environments (Figs. S2 and S3).However, type I error may occur with such a low threshold.Thus, to both detect a set number of true positive loci and reduce the false-positive frequency, we still selected the threshold of 4.2 for HN and 4.6 for HL, although only one QTL was identified in all environments, thereby providing more accurate genetic loci for maize molecular breeding.
Epistasis is interaction between nonallelic genes whereby one gene interferes with phenotypic expression of another gene [42], and has been suggested [43–45] to play an important role in expression of quantitative traits of maize. For HN, the total phenotypic variance explained by additive QTL was 41.1%, and the total contribution of the AA was 43.8% (Tables S4 and S5), indicating that AA was as important to HN as A.For HL, the total phenotypic variance explained by additive QTL was 39.3%, whereas the total contribution of the AA was 54.1%, indicating that AA played a larger role in HL than A.Most of the additive QTL were not involved in epistatic interaction (Tables S4 and S5), suggesting that many QTL in epistatic interactions may not have significant effects for HN and HL alone but may affect these two traits by epistatic interaction with other loci.
Of the nine stable QTL, four (qHN1, qHN7, qHL3,and qHL4.1)have been described previously [20], whereas the remaining five were putatively novel loci. However, only three QTL (qHN7,qHL4.1, and qHL4.2) accounted for >10% of the phenotypic variation. Considering all known QTL studies of maize HN and HL [6,19,20], nearly 40 genetic loci have been detected by linkage and association analysis, of which 11 QTL explain >10% of phenotypic variation (Table S7). These findings suggest that HN and HL are controlled by a small number of major-effect QTL and some minor-effect QTL, reflecting the genetic complexity of maize husk traits. The five overlapping QTL on chromosome 2,3, 7, and 9 (Table S7), suggest that some QTL had pleiotropic effects on HN and HL [20].
4.4. Fine mapping of qHN7, a major-effect QTL for HN
Generally, the ultimate objective of QTL mapping is to clone genes of interest. Haplotype analysis using high-density markers within a target QTL interval in segregating populations is an effective means of localizing primary QTL with large effects [46,47]. Using this strategy, we reduced the qHN7 interval length from 23.8 Mb to 6.5 Mb (Fig. 3b, c). To further narrow the interval containing qHN7, we developed a large BC1F2segregating population with 2566 plants and six polymorphic markers. As a result, qHN7 was delimited to an interval of 721.1 kb between the InDel markers I22 and I33(Fig. 3d, e), which contains 32 genes according to the B73 RefGen_v3 reference genome (Table S6).
4.5. Candidate gene analysis of qHN7
The growth of the maize husk, like that of other organs, is a dynamic process regulated by gene expression [19]. We consider the four candidate genes known to be involved in plant growth and development to be the most likely qHN7 candidate genes in the 721.1 kb interval (Table S6).GRMZM2G159221 and GRMZM2G159094 are respectively zincfinger and NAC transcription factor genes, which regulate plant organogenesis [48–52]. GRMZM2G040947 encodes a pentatricopeptide repeat-containing protein involved mainly in kernel development [53–55]. GRMZM2G166963 encodes a ribosomal protein. Mutations of ribosomal genes in plants may affect leaf morphology. Examples include RML1 in rice[56], and PGY1, PGY2, PGY3, ae5, GDP1, and OLI2 in Arabidopsis[57–59]. Construction of a larger segregating population,candidate gene association mapping, and functional validation may reveal the target gene.
4.6. Application of qHN7 in maize breeding
Marker-assisted selection (MAS) is an effective method to improve target traits in maize [60]. Liu et al. [61] transferred a major-effect QTL for kernel row number (KNR4) into two maize inbred lines using MAS. Lines carrying favorable KNR4 alleles showed increases in kernel row number by almost two rows. Sarika et al. [62] pyramided two quality protein genes(opaque2 and opaque16) to increase protein quality in maize.The pyramided lines showed grain lysine and tryptophan contents as high as respectively 76% and 91% greater than those of the recurrent parents. In the present study, we identified a stable and major-effect QTL associated with HN,qHN7, and mapped it to a relatively small genetic interval,reducing the probability of linkage drag introducing deleterious alleles. The tightly flanking molecular markers with qHN7 could be used to improve the HN of maize inbred lines,such as Chang7-2,an elite Chinese inbred line with 10 layers of husk [6], by marker-assisted backcrossing or recurrent selection. We propose that qHN7 could be used for improvement of maize husk number and cloning of the huskassociated gene.
5. Conclusions
We identified nine stable QTL, 22 additive QTL, and 46 epistatic QTL associated with HN and HL. Both additive and epistatic effects were important. A few major-effect and some minor-effect QTL were the main contributors to the phenotypic variation of HN and HL, reflecting the genetically complex nature of husk traits. The largest-effect QTL for HN on chromosome 7, qHN7, was delimited to a 721.1 kb physical region and four candidate genes were identified. The results will be useful for improvement of husk traits in maize breeding and for the cloning of qHN7.
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2020.03.009.
Declaration of competing interest
Authors declare that there are no conflicts of interest.
Acknowledgments
This study was supported by the Natural Science Foundation of Jiangsu Province (BK20171252), the Jiangsu Agriculture Science and Technology Innovation Fund (CX(19)3049), the Scientific and Technological Project of Jiangsu Province,(BE2018325), the Six Major Talent Project of Jiangsu Province(2016-NY-143 and 2017-NY-138), and the Earmarked Fund for Jiangsu Agricultural Industry Technology System. We thank Professor Chenwu Xu of the Yangzhou University for kindly providing the genotype of RIL population and the resequencing information for DH4866 and T877.
Author contributions
Guangfei Zhou and Derong Hao conceived the project;Yuxiang Mao, Lin Xue, Guoqing Chen, Huhua Lu, Mingliang Shi, Xiaolan Huang, and Derong Hao developed the RIL population; Guangfei Zhou, Yuxiang Mao, Lin Xue, Guoqing Chen, Huhua Lu, Mingliang Shi, Zhenliang Zhang, Xiaolan Huang, Xudong Song, and Derong Hao conducted the field experiments and collected the phenotypic data; Guangfei Zhou analyzed the genotypic and phenotypic data; Guangfei Zhou and Derong Hao wrote the manuscript.

- The Crop Journal的其它文章
- Application of moderate nitrogen levels alleviates yield loss and grain quality deterioration caused by post-silking heat stress in fresh waxy maize
- Identification of a novel planthopper resistance gene from wild rice(Oryza rufipogon Griff.)
- Genome-wide linkage mapping of QTL for root hair length in a Chinese common wheat population
- Comparative analysis of the photosynthetic physiology and transcriptome of a high-yielding wheat variety and its parents
- Metabolic profiling of DREB-overexpressing transgenic wheat seeds by liquid chromatography–mass spectrometry
- Haplotype variations in QTL for salt tolerance in Chinese wheat accessions identified by markerbased and pedigree-based kinship analyses