
Pmn21结构Li2VSiO4电子结构性质
2020-12-22 06:37谢升伦林传金
闽南师范大学学报(自然科学版) 2020年4期
谢升伦,林 硕,林传金
(闽南师范大学物理与信息工程学院,福建漳州363000)
锂离子电池是一种高效率的能源存储与转化装置,具有安全、高能量密度以及使用寿命长等特点,一直在可移动储能方面占有重要的市场地位.目前,锂离子电池主要应用于便携式电子设备,诸如手机、数码相机、笔记本电脑等电子设备的电源.甚至,开始尝试将锂离子电池制备成为电动汽车的能源装置.随着科学创新的不断发展,人们需要能量密度更高、性能更优越,安全性更高且成本低廉的锂离子电池[1].因而开发和探索这种新型锂离子电池是目前的主要方向.在过渡金属中,Fe基锂离子电池因为原料丰富,一直是研究的热门,主要涉及以下几个体系:LiFeO2、LiFePO4、Li2FeSiO4等.LiFePO4材料做成的锂离子电池工作电压相对稳定,比容量也较高,为170 mAh/g,热稳定性和循环稳定性很好,安全性好[2].但是LiFePO4的电子电导率较低,约为10-9~10-6S/cm,因此导电性略差,Li+的扩散系数同样也不高,约为1.8×-14cm2/s[3].Li2FeSiO4分子材料最多只有一个Li 离子可提供脱出/嵌入,根据相关的研究报道,其比容量理论值为166 mAh/g.如果能在实验,或者甚至在实际应用上能实现2 个锂离子的脱出/嵌入,其比容量理论值可达332 mAh/g.实际上,由于Li2FeSiO4自身的电子电导率低,锂离子扩散系数小等缺点,导致该材料的可逆容量低、在常温环境下的电化学性能差、循环倍率性能较低[2].已有实验研究发现Li2FeSiO4材料在掺杂过渡金属V 情况下,材料导电性能得到改善.并且在脱Li+过程中,也能保持结构稳定性[4].目前关于Pmn21结构的Li2VSiO4在实验上还没有相关的报道.本文将从理论上研究这种材料的电子结构性质,旨在进一步丰富对此类材料性质的认识,并对此类材料的探索和制备提供了一定的理论依据.
1 计算方法
本文采用基于密度泛函理论的第一性原理计算方法,交换关联泛函部分采用广义梯度近似(generalized gradient approximation, GGA)[5],使用Perdew-Burke-Ernzerhof形式的赝势库[6],计算了Pmn21结 构Li2VSiO4的电子结构.结构优化和电子结构计算时的Brillouin 区积分采用了Monkhorst-Pack 特殊k 点取样方法,k点网格采用6×4×2.经过反复测试筛选后,平面波截断动能选取520 eV,系统总能量的收敛标准为1×10-5eV/atom.
2 模型构建与结果分析
本文讨论的Li2VSiO4材料属于对称性为Pmn21的正交晶系,标记为Pmn21-Li2VSiO4,采用2×2×2 超晶胞进行研究,晶体结构如图1所示.空间群Pmn21结构的特点:VO4四面体和SiO4四面体交替排布,并共点连接组成[VSiO4]褶皱层,LiO4四面体位于两个[VSiO4]层之间.LiO4四面体中,前3 个O2-来自于同一层[VSiO4],第4个O2-处在另一层[VSiO4].
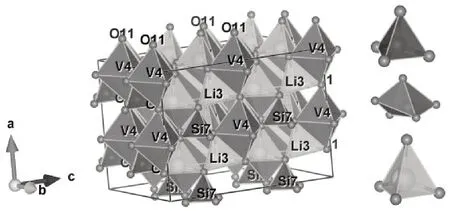
图1 正交结构Li2VSiO4晶体模型Fig.1 Crystal model of Li2VSiO4 with orthogonal structure
2.1 结构稳定性和电荷密度图
为了形成对比,我们也计算了同样结构的Li2FeSiO4.计算得到的两种材料的结构参数由表1给出.我们可以发现,对比Li2FeSiO4在a、b方向上晶格常数呈现缩小而c轴方向呈现一定的膨胀,但是计算得到的总体积比较小;对Li2VSiO4而言,Si-O 键的键长较小于Li-O 和V-O 键,由此可见,Si-O 键的相互作用力较强,可能是共价键.另外,Li-O和V-O键的相互作用力较弱,可能是离子键.
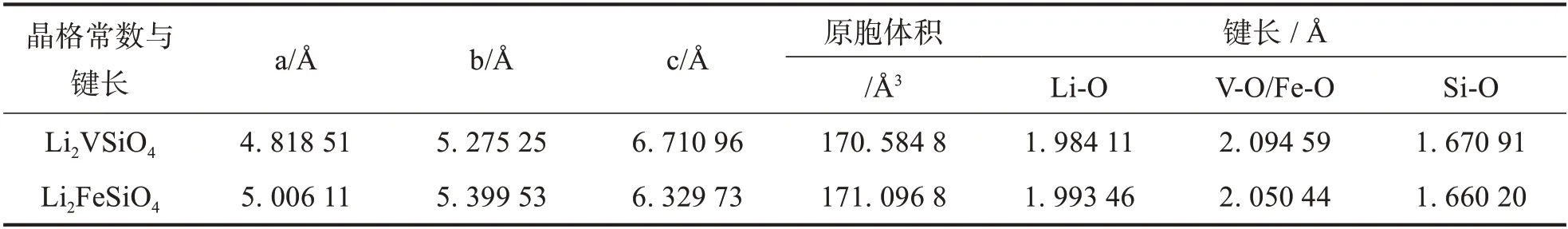
表1 Pmn21-Li2VSiO4和Pmn21-Li2FeSiO4的结构参数与键长Tab.1 Structural parameters and bond length of Pmn21-Li2VSiO4 and Pmn21-Li2FeSiO4
图2是计算的电荷密度图,在分析图2(a)后可确定,Si-O 键具有稳定的共价键性质(电荷偏球形分布体现了共价性),可使SiO4四面体结构可以良好的维持Li2VSiO4主体框架.在分析图2(b)后,可知V-O键是离子键(电荷球形分布体现离子键).为了更好的研究V-O 键电子得失情况,图3给出了差分电荷密度图(可以清楚地得到反应过程中,电荷移动和电荷成键极化方向等性质),差分电荷密度计算公式为:其中ρ(r)是体系的总电荷密度,而是体系中所有独立原子电荷密度之和.从图3中可以看出,V-O的电荷转移非常明显(V失去电子,O得到电子).因此,Li2VSiO4材料的电子键合特点是离子性和共价性的混合.
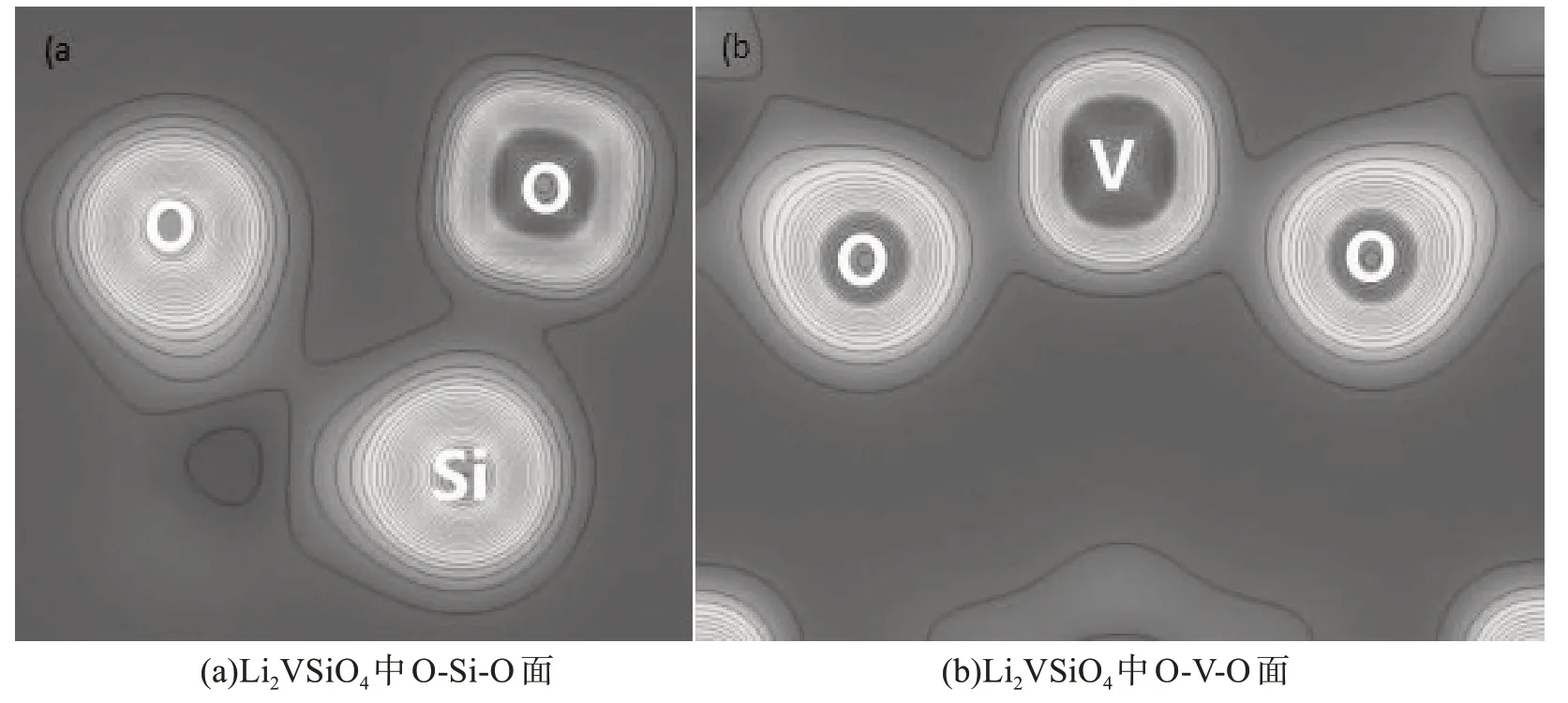
图2 电荷密度图Fig.2 Charge density

图3 Li2VSiO4差分电荷密度图Fig.3 Differential charge density of Li2VSiO4
2.2 能带结构和态密度分析
图4分别给出计算得到的Li2VSiO4材料自旋向上(spin-up)和向下(spin-down)的能带结构图.由导带和价带的能带间隙可得到,Li2VSiO4的spin-up 的带隙约1.75 eV,自旋向下的带隙约5.1 eV,费米能级都处在价带与导带之间,均符合半导体费米能级处在价带与导带之间的性质,但是spin-down 的带隙较大,接近绝缘体的性质,由此可见,spin-up 和spin-down 能带性质相差比较大.图5、图6是计算得到的Li2VSiO4材料的总态密度图和各个原子的分波态密度图.根据态密度图可以清楚地得到结论,在费米能级附近,V-3d态轨道电子对态密度贡献最为显著,其次是O-2p态轨道电子.在spin-up的导带中,Li-2s态、Si-3s 态和Si-3d 态电子贡献相对较弱,而V-3d 态和O-2p 态电子贡献相对较强.Spin-up 的价带中,在-5.0~2.8 eV 范围内,O-2p 态轨道电子对态密度的贡献最大,其次是V-3d 态电子.在spin-down 的导带中,Li-2s态、Si-3s态和Si-3p态电子的贡献比spin-up时略高,但总体贡献还是偏低.V-3d态和O-2p态电子贡献也比spin-up 时高,其中V-d 态电子远高于spin-up 时的态密度,这也是spin-down 能带在4.5 eV 附近较平缓的原因.Spin-down的价带和spin-up相差不大,唯一的区别是,在-2.6~0 eV范围内,没有电子态密度.因此,Li2VSiO4材料spin-up时,费米能级附近可利用的态密度更多,导电性更好,Li2VSiO4材料整体上呈现半导体性质.
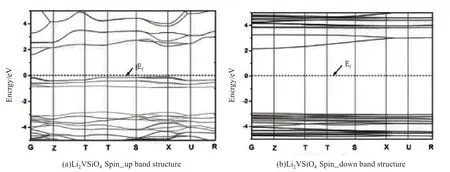
图4 (a)Li2VSiO4自旋向上能带结构(b)Li2VSiO4 自旋向下能带结构Fig.4 (a)Spin up band structure of Li2VSiO4(b)Spin down band structure of Li2VSiO4
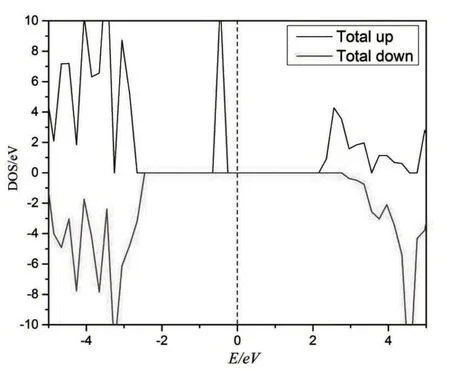
图5 Li2VSiO4总态密度图Fig.5 Total density of states of Li2VSiO4
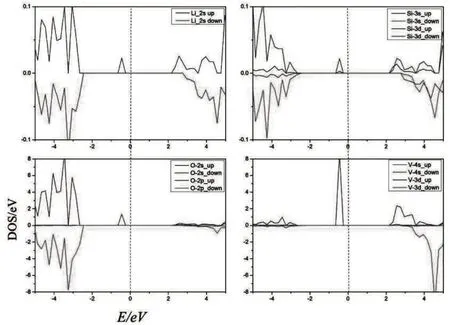
图6 Li、V、Si、O分波态密度图Fig.6 Partial density of states of Li、V、Si、O
3 结论
本论文采用基于DFT 的第一性原理方法计算了锂电池正极材料Li2VSiO4,并进行结构优化,得到晶体的结构参数,与Li2FeSiO4进行对比,Li-O 键长较小,V-O 键长略大,Si-O 键长略大,单位晶胞体积较小;计算的电荷密度和差分电荷密度,结果表明Li2VSiO4材料的电子键特点是离子性和共价性的混合;计算了Li2VSiO4的电子结构,从能带图来看,Pmn21-Li2VSiO4材料具有半导体材料的性质,计算spin-up和spindown 的能带的带隙分别为1.75 eV 和5.1 eV,自旋极化能带特性相差较大,其在自旋材料中有一定的应用价值.因此对Li2VSiO4材料的研究具有重要的理论意义和应用参考价值.
猜你喜欢
装备维修技术(2021年36期)2021-10-25
小天使·聪聪画刊(2021年2期)2021-09-10
弹箭与制导学报(2021年3期)2021-07-30
汽车零部件(2020年10期)2020-11-09
原子与分子物理学报(2020年3期)2020-05-15
汉语世界(The World of Chinese)(2019年6期)2019-09-10
网印工业(2019年4期)2019-05-21
重型机械(2019年2期)2019-04-28
西安工业大学学报(2019年2期)2019-04-02
吉首大学学报(自然科学版)(2018年3期)2018-07-03
