
自噬调控因子mTOR对心肌缺血再灌注损伤的作用机制研究进展
2020-12-17 07:56:16韩利民罗小红赵海龙
解放军医学杂志 2020年11期
韩利民,罗小红,赵海龙*
1遵义医科大学基础医学院病理生理学教研室,贵州遵义 563000;2重庆市长寿区人民医院病理科,重庆 401220
缺血性心脏病已成为我国居民死亡的第二大病因,临床上采用经皮冠状动脉介入治疗(percutaneous coronary intervention,PCI)可有效地改善心肌灌注,是降低患者病死率的重要措施[1]。 但心肌缺血后血流再灌注常会对心肌产生二次损伤,该过程被称为心肌缺血再灌注(ischemia/reperfusion,I/R)损伤。据统计,美国每年约有66万人因初发急性心肌梗死死亡,约30万人因再发心肌梗死死亡[2]。探索更多治疗PCI术后心肌I/R损伤的策略仍然是现阶段亟待解决的问题之一。
自噬是细胞通过单层或双层膜包裹待降解物形成自噬体,然后运送到溶酶体形成自噬溶酶体,并进行多种酶消化及降解的过程[3-4]。多项证据表明,自噬参与了心肌I/R损伤过程,自噬不足或过度均是引起心肌I/R损伤的重要原因,但其具体机制尚不完全清楚[5-7]。哺乳动物雷帕霉素靶点(mammalian target of rapamycin,mTOR)是自噬的重要调控因子,已被证实与肿瘤、神经系统疾病、代谢性疾病及心血管疾病等多种疾病息息相关[8-14]。近年来,有关心肌I/R损伤的体内及体外研究表明,多种药物均可通过mTOR调控自噬,从而在一定程度上改善心肌I/R损伤[6,15-18]。本文对mTOR在心肌I/R损伤中的研究进展进行综述,旨在明确自噬调控因子mTOR改善心肌I/R损伤的相关作用机制,以期为治疗心肌I/R相关药物的研发提供依据。
1 mTOR简介
1.1 mTOR的结构 mTOR是哺乳动物中雷帕霉素的直接靶点,属于磷脂酰肌醇-3激酶相关激酶(phosphatidylinositol-3-kinase PI3K related protein kinase,PIKK)家族成员。mTOR结构包括氨基端20个串联重复序列,羧基端的蛋白激酶结构域,以及位于激酶上游的FRB、FAT两个结构域和下游的FATC结构域[19]。mTOR与不同适配器蛋白结合可形成mTORC1及mTORC2两种复合物。其中,mTOR调控蛋白(regulatory protein associated with mTOR,raptor)是mTORC1的核心元件之一,主要负责底物的募集;mLST8是其另一个核心元件,可激活激酶结构域;40 ku大小的富含脯氨酸蛋白激酶B底物蛋白(proline-rich AKT substrate 40,PRAS40)、DEP结构域含有雷帕霉素靶蛋白相互作用蛋白(DEP domain-containing mTOR interacting protein,deptor)与raptor作用,发挥负向调节mTORC1的功 能[20]。而mTORC2的活性区域是mTOR的雷帕霉素不敏感伴侣(rapamycin-insensitive companion of mTOR,rictor),其功能是与底物结合;protor 1/2、mSin1通过与rictor结合以维持mTORC2的稳定及活化[21]。
1.2 mTOR的生物学功能 mTOR是细胞生长发育的关键因子,在维持机体正常的生物学功能中起重要作用。Potter等[22]的研究证实,采用不同剂量的mTOR抑制剂雷帕霉素喂食果蝇幼虫可导致其出现行为缺陷及发育迟缓。Sciarretta等[20]研究发现,mTOR基因敲除(mTOR-KO)的新生小鼠心肌细胞发育不良,在出生3周后可迅速出现扩张型心肌病导致死亡。p70S6K是mTOR的下游基因,可控制细胞生长、增殖及分化等过程,mTOR可直接促使其磷酸化,从而使p70S6K激活[23]。同时,mTOR也可激活真核起始因子4F(eukaryotic translation initiation factor 4F,eIF4G),并抑制4EBP1/2增强mRNA转录,促进蛋白质合成[24]。此外,mTOR还可直接激活胆固醇调节元件结合蛋白(sterol regulatory element binding protein,SREBP)及过氧化物酶体增殖物激活受体γ(peroxisome proliferator activated receptor γ,PPARγ),同时间接抑制Lipin1的表达,进而促进脂质合成[25]。
2 mTOR对自噬过程的影响及其调控机制
自噬过程通过溶酶体对非功能性蛋白进行降解,可促使营养物质被机体重新利用,因此曾被看作是机体在各种应激条件下(如缺血、缺氧等)的一种自我保护反应[26]。mTOR是自噬的重要负性调控因子,可通过抑制自噬复合物ULK-ATG13-FIP200形成,从而抑制自噬过程的激活[27]。AMPK/mTOR及PI3K/AKT/mTOR是调控自噬的两条重要信号通路,前者在能量不足时被激活,而后者在感受到胰岛素等生长因子后被激活。Zeng等[28]发现,在心肌缺血阶段,AMPK/mTOR信号通路被激活,电镜下可观察到心肌细胞自噬小体数量明显增加。Wang等[29]发现,与正常大鼠相比,糖尿病性脑病大鼠模型中PI3k/AKT/mTOR信号增加,使自噬水平下降。由此可见,不同条件下mTOR介导的自噬在维持机体的稳态中均发挥着重要作用。
3 mTOR对心肌缺血及再灌注阶段的不同作用及其机制
自噬在心肌I/R损伤中具有双重作用,适度自噬可促进细胞存活,而过度自噬则会引起细胞发生自噬性死亡。mTOR在不同阶段可通过不同机制调控自噬水平,减轻心肌细胞的损害。
3.1 mTOR在心肌缺血阶段的作用及机制 Lin等[15]研究发现,在心肌缺血阶段,mTOR因能量不足及缺氧被抑制,从而激活自噬过程,促进心肌细胞存活。AMPK/mTOR是该过程诱导自噬激活的主要通路,心肌能量不足时,AMP/ATP上升,糖原合成酶激酶-3β(glycogen synthase kinase 3β,GSK-3β)也被激活,两者可直接激活结节性硬化复合物1/2(tuberous sclerosis complex 1/2,TSC1/2),抑制下游mTOR蛋白的表达。此外,还有其他机制参与抑制mTOR:①心肌能量不足及缺氧可分别激活p38调节活化蛋白激酶(p38-regulated/activated kinase,PRAK)、B淋巴细胞瘤-2基因/腺病毒E1B相互作用蛋白3(Bcl-2/adenovirus E1B 19-kD protein-interacting protein 3,BNIP3),导致Rheb(脑中富含的Ras同源蛋白)失去结合GTP的能力,阻断mTOR的激活[30]; ②心肌缺氧诱导产生的缺氧诱导因子α(hypoxia inducible factor-1α,HIF-α)激活发育及DNA损伤调节基因1(regulated in DNA damage and development 1,REDD1)后通过TSC1/2间接抑制mTOR[20](图1)。
3.2 mTOR在再灌注阶段的作用及机制 Shi等[5]发现,再灌注阶段心肌细胞自噬水平明显升高,其原因与Beclin 1的激活有关,而与AMPK/mTOR无关。具体机制如下:①该过程产生大量的活性氧(reactive oxygen species,ROS)通过ROS-JNK信号通路干扰Beclin 1/Bcl-2的相互作用,使Beclin 1从Bcl-2中游离出来,从而促进自噬的发生[31];②该过程的Ca2+超载促使钙调素依赖性蛋白激酶Ⅱ(calcium/calmodulin-dependent protein kinase Ⅱ,CaMKⅡ)在Ser90位点直接磷酸化Beclin 1,促进K63连接的Beclin 1泛素化并激活自噬[32]。以上机制最终使心肌细胞发生过度自噬,导致细胞死亡。此时,mTOR因血流恢复及炎性因子被激活,在一定程度上可抑制过度自噬,抵抗心肌I/R损伤。一方面,心肌血流恢复后,PI3K/AKT信号通路在激素、生长因子等的刺激下被激活,PRAS40对mTOR的抑制作用解除,同时还能抑制TSC1/2间接激活mTOR,最终抑制自噬过程。另一方面,大量ROS可触发炎症反应,通过IKKβ/NF-κB信号通路抑制TSC1/2,最终导致mTOR被激活,从而抑制自噬[33](图1)。Zeng等[28]发现,在心肌I/R阶段,与对照组相比,西红花苷处理可明显减少心肌细胞凋亡,电镜下心肌细胞自噬小体的数量明显减少,其机制与AKT/mTOR信号通路激活有关。
4 基于mTOR的相关信号通路与心肌I/R损伤
mTOR作为自噬的重要调控因子,越来越多的证据表明,多种药物可作用于mTOR相关信号通路,调控不恰当的自噬水平,从而改善心肌I/R损伤,mTOR可能是治疗心肌I/R损伤的潜在靶点。
4.1 PI3K/AKT/mTOR信号通路与心肌I/R损伤 PI3K/AKT/mTOR是一种调控自噬的经典信号通路,与心血管疾病密切相关。Lin等[34]研究证实,促红细胞生成素衍生肽即螺旋B表面肽(helix B surface peptide,HBSP)可通过激活PI3K/AKT/mTOR信号通路减轻心肌I/R诱导的过度自噬及细胞凋亡。Zhang等[35]发现,远程缺血后处理在体内及体外均有对抗心肌I/R损伤的心脏保护作用,其机制与PI3K/mTOR信号通路上调乙醛脱氢酶2(ALDH2)的表达有关。此外,多项研究表明,多种药物均可通过作用于PI3K/AKT/mTOR信号通路而抑制细胞过度自噬,在一定程度上减轻心肌I/R损伤(表1)。
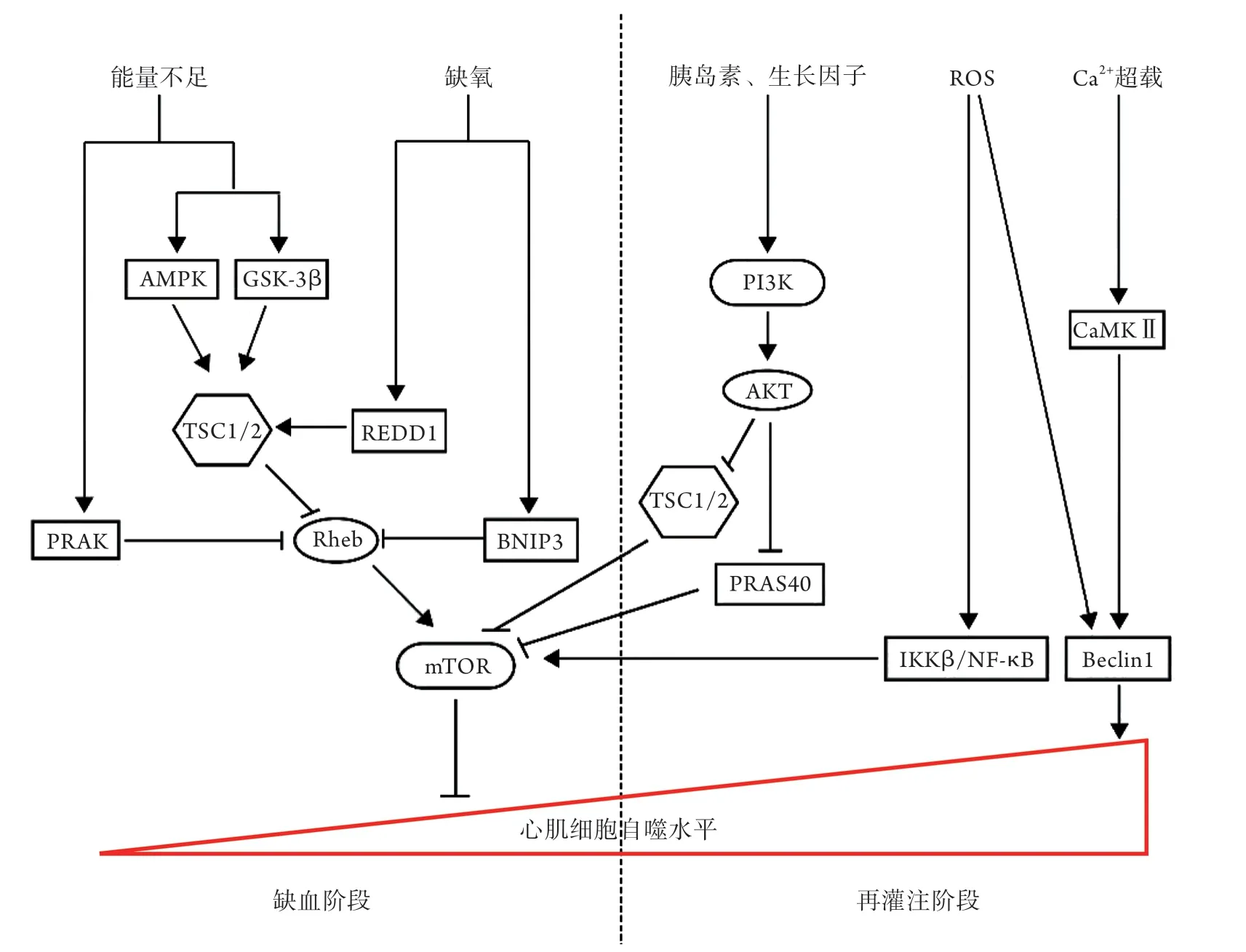
图1 mTOR对心肌缺血再灌注损伤的作用机制Fig.1 Mechanism of mTOR in myocardial ischemia-reperfusion injury
4.2 AMPK/mTOR信号通路与心肌I/R损伤AMPK是mTOR的上游抑制因子,对能量不足尤为敏感。Fu等[6]的研究表明,与对照组相比,天麻素预处理可通过增加自噬通量(p62蛋白水平下降、LC3Ⅱ蛋白水平增高)减轻小鼠心肌I/R损伤,其机制与p-AMPK升高及p-mTOR降低有关;加入AMPK特异性抑制剂后小鼠心肌自噬水平降低,心功能恶化,表现为左室射血分数(left ventricular ejection fraction,LVEF)、左室短轴缩短分数(left ventricular fraction shortening,LVFS)均明显下降。Yang等[7]发现,一种分泌型脂肪因子丝氨酸蛋白酶抑制剂(vaspin)可依赖AMPK/mTOR的激活而上调自噬通量,抑制心肌细胞凋亡,而使用氯喹抑制自噬通量后则降低了vaspin的保护作用。此外,一项临床研究证实,对PCI术后患者予以黄连素治疗后,血浆C反应蛋白(CRP)、TNF-α及IL-6水平较对照组均明显降低,考虑与AMPK/mTOR激活有关[36]。综上,AMPK/mTOR信号通路激活可提高细胞自噬水平,从而改善心肌I/R损伤。
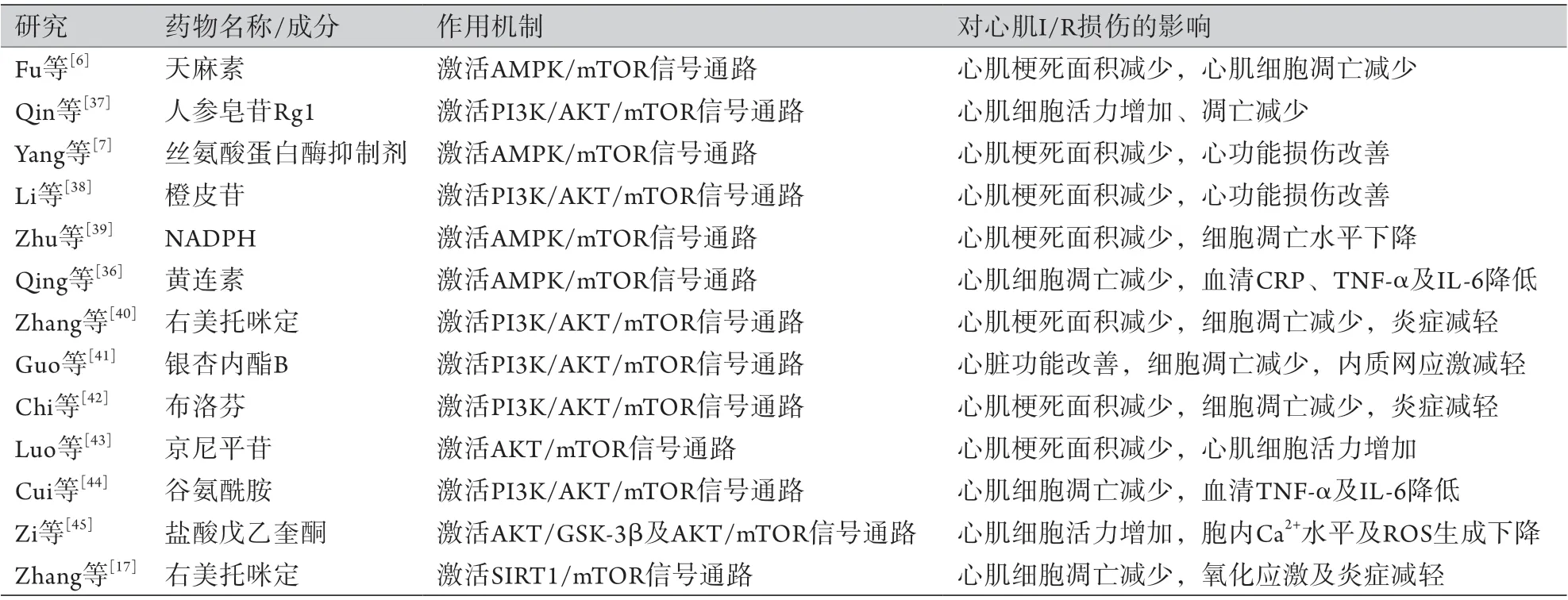
表1 mTOR与心肌I/R损伤的相关研究Tab.1 Related researches on mTOR and myocardial I/R injury
4.3 其他mTOR信号通路与心肌I/R损伤 除PI3K/AKT/mTOR及AMPK/mTOR两条重要信号通路外,还存在其他mTOR通路与心肌I/R损伤有关。Zhang等[17]发现,右美托咪定可减轻大鼠I/R后的心肌细胞凋亡、氧化应激及炎性反应,其机制与SIRT1/mTOR信号通路激活有关。Li等[46]研究证实,骨髓间充质干细胞衍生的条件培养基可通过激活Notch2/mTOR信号通路降低H9C2细胞缺氧/复氧后的自噬水平,增强H9C2细胞活力,并减少ROS的产生。
5 总结与展望
目前临床上对于PCI术后血流再灌注引起的心肌I/R损伤并无特效药物,主要是对其机制进行干预。自噬调控因子mTOR在改善心肌I/R损伤中发挥重要作用。心肌缺血阶段,AMPK/mTOR通路的激活可增强自噬,促进心肌细胞存活;而再灌注阶段,PI3K/AKT/mTOR通路的激活可通过抑制过度自噬而减少心肌细胞死亡。因此,PCI术后使用药物或其他方式激活mTOR可能是预防及治疗心肌I/R 损伤的策略之一。此外,基于mTOR在心肌缺血阶段的作用机制,对于仅合并轻度心肌缺血暂无PCI手术指征的大部分临床患者,是否可通过抑制mTOR途径减少缺血对心肌细胞的损伤尚需进一步探讨。
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25 13:16:04
世界科学技术-中医药现代化(2021年7期)2021-11-04 08:10:24
学苑创造·A版(2020年12期)2020-01-07 14:07:23
中国外汇(2019年15期)2019-10-14 01:00:34
广州大学学报(自然科学版)(2019年1期)2019-05-07 01:33:26
作文教学研究(2016年1期)2016-07-05 12:22:47
海南医学(2016年8期)2016-06-08 05:43:00
天津科技大学学报(2016年1期)2016-02-28 16:59:45
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10 08:41:53
中国病理生理杂志(2015年8期)2015-12-21 12:38:08
