
TGF-β信号通路在纤维化疾病中的作用研究进展
2020-12-17 07:56:16范美玲应苗法赵蕊金烨成李明星
解放军医学杂志 2020年11期
范美玲,应苗法,赵蕊,金烨成,李明星
浙江大学医学院附属邵逸夫医院药剂科,杭州 310016
纤维化是许多慢性炎性疾病的最终病理结果,其主要病理特征是炎症或受损组织周围的纤维结缔组织过度聚积,引起细胞外基质(extracellular matrix,ECM)中的胶原蛋白及纤连蛋白过度增加,成纤维细胞向肌成纤维细胞分化,导致永久性瘢痕、器官衰竭甚至死亡[1-3]。临床上常见的纤维化疾病包括肺、肝、心肌、肾、骨髓纤维化及硬化病等[4-5],尽管不同纤维化疾病的发病原因和临床表现各不相同,但其最终的致病机制是一致的。转化生长因子-β(transforming growth factor-β,TGF-β)是纤维化疾病发生的重要调控因子,能够调控成纤维细胞向肌成纤维细胞转化,通过与下游细胞质转录因子Smads作用,诱导胶原的合成[6-7]。因此,深入研究TGF-β在纤维化疾病中的作用及其调控机制,对纤维化疾病的治疗尤为重要。本文将对TGF-β信号通路在纤维化疾病发生机制中的研究进展进行综述,以期为新的靶向药物的开发提供理论依据。
1 TGF-β信号通路的特征及作用机制
1.1 TGF-β信号通路的特征
1.1.1 TGF-β的组成 TGF-β属于细胞因子超家族,后者由TGF-β、活化素、抑制素、生长和分化因子(growth and differentiation factors,GDF)、骨形成蛋白(bone morphogenetic proteins,BMP)等组成,主要分成TGF-β-活化素亚家族和BMP亚家族。TGF-β具有二聚体结构,在所有细胞中均表达,在调控细胞的生长、迁移、免疫抑制、内皮间质转换及ECM重构等方面具有重要作用[8]。已发现的TGF-β有5个亚型,即TGF-β1-TGF-β5;然而,在哺乳动物中,TGF-β仅包含3个亚型,即TGF-β1、TGF-β2、TGF-β3,各亚型在调控生物功能过程中的作用相似,其中TGF-β1在各组织中的表达水平最高,活性最强,广泛参与胚胎发育、组织纤维化及肿瘤的发生发展等生理病理过程。
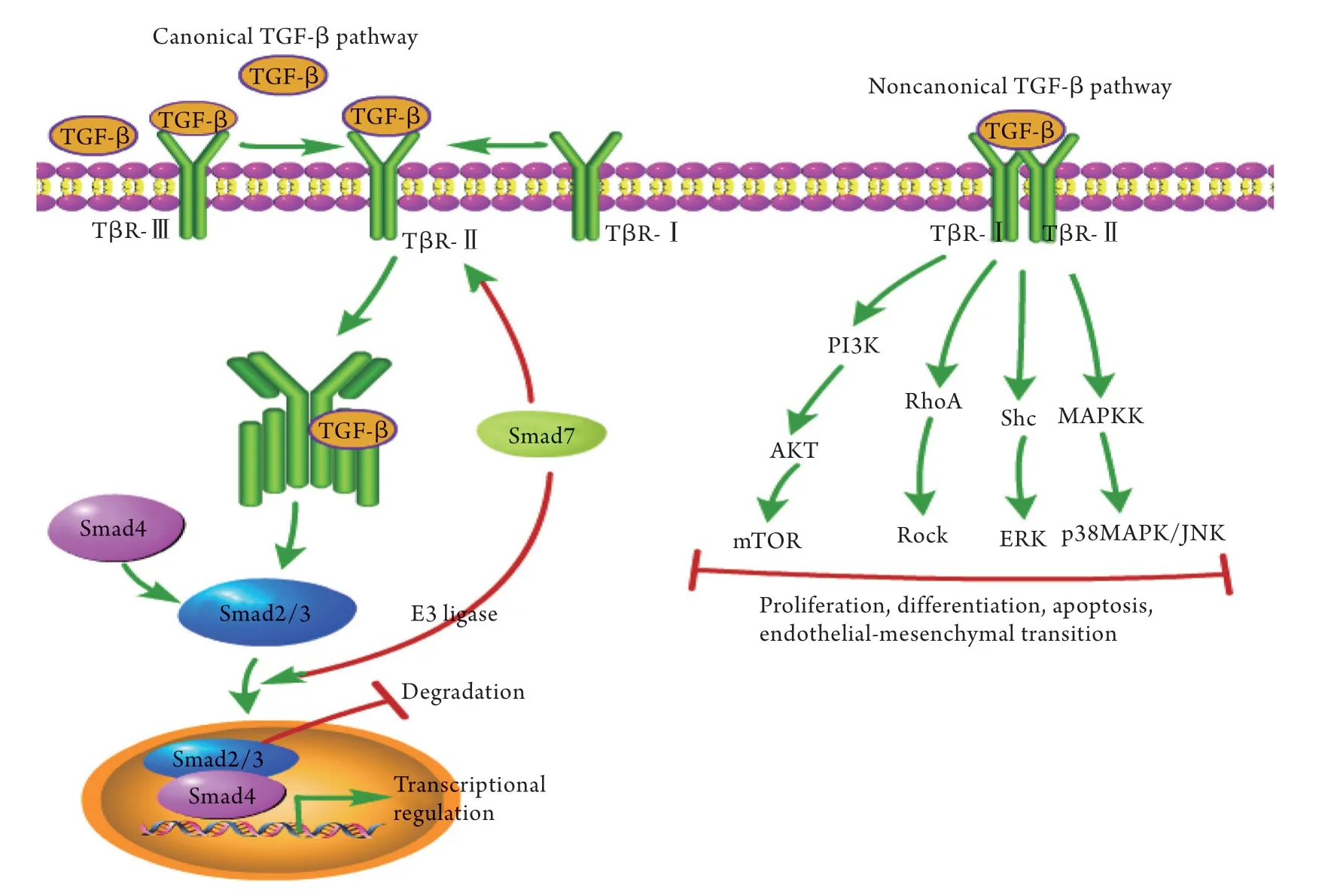
图1 TGF-β信号通路的作用机制Fig.1 The mechanisms of TGF-β signaling pathway
1.1.2 TGF-β受体的分类及信号传递 TGF-β受体包含3个亚家族,即Ⅰ型受体(type Ⅰ TGF-β receptor,TβR-Ⅰ)、Ⅱ型受体(TβR-Ⅱ)及Ⅲ型受体(TβR-Ⅲ),其受体与配体具有类似的生物活性,均参与调控细胞的增殖、分化及凋亡等过程[9]。有研究证实,TβR-Ⅰ不能单独与TGF-β结合发挥作用,需要TβR-Ⅱ共同参与,而TβR-Ⅱ可直接与TGF-β1结合;TβR-Ⅲ与TGF-β1具有较高的亲和性,其主要功能是将TGF-β传递至受体分子,与其他两个受体共同参与信号的传递[10]。Smads蛋白是TGF-β下游的信号蛋白,能够将TGF-β信号从细胞外传递至细胞核发挥生物学功能。根据其结构及功能,Smads蛋白可分为3种不同的亚型:受体调节型Smads蛋白(包括Smad1/2/3/5/8)、共同调节型Smads蛋白(Smad4)及抑制型Smads蛋白(包括Smad6及Smad7);其中Smad2及Smad3参与了TGF-β信号的传递,Smad1/5/8参与了BMP信号的传递[11]。
1.2 TGF-β信号通路的作用机制 TGF-β信号通路主要分为经典的TGF-β/Smads信号通路和非经典的TGF-β信号通路,其信号通路作用的过程如图1所示,具体的作用机制如下。
1.2.1 经典的TGF-β/Smads信号通路 正常情况下,TGF-β主要存在于ECM中,处于失活状态。在经典的TGF-β/Smad信号传递过程中,配体先与TβR-Ⅱ结合,并将TβR-Ⅰ、TβR-Ⅲ招募至ECM,磷酸化TβR-Ⅰ并与其结合,形成TβR-Ⅰ-TGF-β-TβR-Ⅱ-TβR-Ⅲ四聚体;磷酸化的TβR-Ⅰ将Smad2及Smad3在丝氨酸残基的C末端磷酸化,招募Smad4并与其形成复合物,转移至细胞核,调控TGF-β靶基因的转录[12]。此外,抑制型Smad7可被招募至TGF-β受体或Smad2/3的复合物中,通过Smad特异性E3连接酶启动其降解,抑制TGF-β的信号转导。
1.2.2 非经典的TGF-β信号通路 除了Smads介导的典型TGF-β信号通路外,TGF-β受体还可以通过信号转导中间体的磷酸化或与其直接相互作用来激活其他细胞内通路,如磷脂酰肌醇激酶/蛋白激酶B(phosphatidylinositol 3-kinases/protein kinase B,PI3K/AKT)通路、细胞外信号调节激酶(extracellular signal regulated kinase,ERK)、氨基末端激酶(c-jun N-terminal kinase,JNK)、丝裂原活化蛋白激酶(mitogen activated protein kinases,MAPK)及RhoA/Rho激酶(RhoA/Rho kinase,RhoA/Rock)信号通路等,被称为非经典的TGF-β信号通路[13]。研究发现,TGF-β与TβR-Ⅰ及TβR-Ⅱ结合后,通过与PI3K亚基p85作用活化PI3K/AKT通路,然后AKT活化下游的哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR),调控基因的转录过程;此外,TGF-β活化后可激活下游的ERK、p38MAPK/JNK及Rock信号通路,参与调控细胞的增殖、分化、凋亡及内皮间质转化[14]。
2 TGF-β信号通路在纤维化疾病发生中的作用机制
2.1 肺纤维化 肺纤维化是由各种致病因素引起的一种复杂的间质性肺病,临床表现为肺组织中存在瘢痕,肺泡壁增厚,可使血氧交换减少,多数患者因肺衰竭发生窒息性死亡[15]。目前认为,导致肺纤维化发生的原因包括环境因素、放射治疗、药物因素及自身免疫性低等。肺组织损伤与修复的异常是肺纤维化形成的主要过程,其病理机制是肺血管内皮细胞及肺泡上皮细胞受到致病因子损伤后,引起肺泡炎症及肺泡反复性损伤,成纤维细胞异常增殖并转化为肌成纤维细胞,胶原蛋白表达增加,ECM大量聚积,进而引起肺组织进行性瘢痕形成,最终导致肺纤维化病变[16]。近年来研究发现,TGF-β是促纤维化发生的主要细胞因子,肺组织中的多种细胞如巨噬细胞、中性粒细胞、肺泡上皮细胞、肌成纤维细胞等均可分泌TGF-β;TGF-β信号通路参与了肺纤维化的发生及发展[17]。
肺纤维化的形成过程涉及经典的TGF-β/Smads信号通路及非经典的TGF-β信号通路。体外研究发现,博来霉素及二氧化硅均可通过活化TGF-β1/Smads信号通路而诱导肺纤维化的形成;安洛替尼及米诺地尔均可抑制博来霉素诱导的TGF-β1/Smad3信号通路活化,抑制上皮间质转化及成纤维细胞的增殖,增加细胞凋亡;丹参酮ⅡA可抑制二氧化硅诱导的TGF-β1/Smad3信号通路活化,上调Smad7的表达,改善肺纤维化[18-20]。另有研究发现,辐射能够诱导TGF-β1的表达,增加胶原蛋白的沉积,诱发肺纤维化;异甘草酸镁可抑制TGF-β1的升高,抑制下游的p38MAPK/AKT/Nox4通路,抑制成纤维细胞的分化,减轻肺纤维化[21]。目前肺纤维化尚无疗效确切的治疗药物,已上市的吡非尼酮(pirfenidone)能够靶向TGF-β,发挥抗肺纤维化的作用,减缓疾病的进展并延长患者的生存时间。深入探究TGF-β信号通路在肺纤维化发生过程中的作用,可为肺纤维化靶向治疗药物的研发提供新的 方向。
2.2 肝纤维化 肝纤维化是一种因机体对各种肝脏损伤修复异常引起的慢性肝脏疾病,并非单一性疾病,多种致病因素如病毒感染、乙醇及药物损伤、脂肪肝、自身免疫性疾病等均会引起肝纤维化,并可进一步发展成肝硬化、肝癌。其主要的病理机制是ECM合成增多,并过量聚积;肝星状细胞(hepatic stellate cell,HSC)活化并分化为肌成纤维细胞。有研究发现,多种信号通路参与了肝纤维化的发生,其中在正常肝组织中表达较低而在肝组织受损时被激活且表达增加的TGF-β被认为是肝纤维化发生的关键因子[22]。在正常的肝组织中,HSC处于非增殖的静止状态;急性或慢性肝损伤后,ECM中的TGF-β被激活并高表达,从而诱导HSC活化,并向增殖、迁移表型转化,促进胶原及ECM蛋白的合成[23]。因此,TGF-β信号参与了HSC活化及胶原的合成,从而调控肝纤维化的进程。
TGF-β1的表达与ECM的聚积相关,在肝纤维化的发生中具有重要作用;Smad3及Smad4是TGF-β1下游的促纤维化基因,Smad2及Smad7可抑制纤维化的发生,敲除Smad3可抑制Ⅰ型胶原蛋白的表达及上皮-肌成纤维细胞转化;Smad4可增强Smad3响应性启动子活性,促进纤维化形成,而Smad7则可负向调控Smad3诱导的纤维形成[24]。在肝纤维化大鼠模型中,TGF-β/Smad/MAPK信号通路被激活,羟基脯氨酸水平及α-肌动蛋白表达均上调,胶原蛋白合成增加,厚朴酚可抑制TGF-β/Smad/MAPK信号通路的活化,有效逆转肝损伤指标的改变;酪氨酸激酶抑制剂尼洛替尼可降低TGF-β1水平,抑制炎性因子的释放,抑制四氯化碳诱导的肝纤维化,发挥潜在的抗纤维化作用[25]。此外,在人源及鼠源纤维化肝组织中,双加氧酶Tet家族蛋白Tet3表达增加,促进TGF-β1的表达上调,诱导HSC转分化为肌成纤维细胞,抑制Tet3/TGF-β1信号通路可减轻小鼠肝纤维化[26]。因此,深入研究TGF-β/Smad信号通路上下游的调控机制有助于更好地设计TGF-β靶向抗肝纤维化药物,但由于TGF-β的生物学功能广泛,目前发现的靶向药物不良反应显著,已超出了治疗获益。
2.3 心肌纤维化 心肌纤维化是指各种致病因素引起的心肌成纤维细胞异常增殖,胶原蛋白合成增加,ECM过度生成及沉积,心脏间质重构的病理过程[27]。心肌纤维化的发病机制较复杂[28],目前研究认为TGF-β信号通路活化、肾素-血管紧张素-醛固酮系统、基质金属蛋白酶(matrix metalloproteinases,MMPs)及基质金属蛋白酶组织抑制剂(tissue inhibitors of matrix metalloproteinase,TIMPs)表达失衡、自噬等参与了心肌纤维化的形成[29]。在心肌细胞中,心肌成纤维细胞约占90%;当心肌成纤维细胞处于静息状态时,分泌的MMPs能够降解ECM,而TIMPs可抑制MMPs的活性,MMPs/TIMPs处于动态平衡;当成纤维细胞受到刺激后可异常增殖,并分化为肌成纤维细胞,使Ⅰ型及Ⅲ型胶原蛋白合成增加,其中,Ⅰ型胶原蛋白主要控制心肌壁的拉伸强度,Ⅲ型胶原蛋白控制心肌的弹性,Ⅰ型/Ⅲ型胶原蛋白比例失衡可导致心肌纤维化的发生[30]。目前研究发现,促纤维生成因子TGF-β在心肌成纤维细胞的增殖、凋亡、分化等过程中发挥重要作用,TGF-β/Smad2/3信号通路参与了心肌纤维化的发生[31]。
在压力负荷诱导的心肌纤维化小鼠模型中,TGF-β受体TβR-Ⅰ、TβR-Ⅱ表达上调,活化了下游的纤维化特异蛋白Smad2/3,从而促进心肌成纤维细胞的分化;敲除TβR-Ⅰ、TβR-Ⅱ或Smad2/3可抑制成纤维细胞的活性,减少ECM相关基因的表达,减轻心肌纤维化并改善心肌细胞的稳态[32]。另外,在高血压伴心肌纤维化大鼠模型中,TGF-β1/Smad2信号通路被激活,参与了心肌纤维化的发生,而淫羊藿苷Ⅱ可逆转该过程[33]。进一步研究发现,非经典的TGF-β信号通路也参与了心肌纤维化的发生;TGF-β活化Wnt/β-catenin信号通路,进而稳定TGF-β/Smad信号通路的活性,TGF-β也可活化下游的p38MAPK、ERK、JNKs等信号分子,调控心肌纤维化的发生[34]。有研究发现,心脏瓣膜纤维化患者的血清TGF-β1表达升高,瓣膜组织中Ⅰ型胶原蛋白及α-平滑肌肌动蛋白(α-smooth muscle actin,α-SMA)表达均升高,p38MAPK、JNK、ERK的表达上调,且TGF-β1的表达与JNK及p38 MAPK的磷酸化呈正相关[35]。另有研究发现,在机械应力损伤小鼠心肌纤维化模型中,辣椒素受体4通过活化TGF-β1/Rho/Rock信号通路调控纤维化基因启动子的活性及成纤维细胞的分化,敲除辣椒素受体4基因可发挥心肌保护作用[36]。因此,经典的及非经典的TGF-β信号通路均参与了心肌纤维化的发生过程,但由于其作用机制较复杂,目前仍未完全阐明。
2.4 肾纤维化 肾纤维化是许多慢性肾病发展的必经阶段,是慢性肾衰竭的最终表现,其主要特征是肾小管间质纤维化、炎性细胞浸润、肾小球硬化及肾实质丧失。目前认为,肾纤维化的病理机制包括炎性细胞浸润、上皮间质转化、成纤维细胞活化、Ⅲ型胶原及纤连蛋白合成增加、ECM过度沉积及肾小管周围微血管堵塞,其中ECM过度沉积是肾纤维化发生的关键因素[37]。长期研究证实,TGF-β1是肾纤维化发生的关键调控因子,可通过激活下游的Smads信号通路诱导肾瘢痕的形成;机体中潜在储存的TGF-β1具有抗炎及抑制肾纤维化发生的作用,而过度活化的TGF-β1则可造成进行性肾损伤[38]。在TGF-β/Smads信号通路中,Smad3具有致病性,Smad2及Smad7具有肾脏保护作用;肾脏损伤时,Smad2及Smad3高度活化,Smad7被泛素-蛋白酶体系统降解;Smad4不仅能够加重Smad3诱导的肾纤维化,还能通过增强Smad7的活性而抑制核因子κB(nuclear factor kappa-B,NF-κB)诱导的肾脏炎症反应[39]。因此,通过Smad7或Smad3依赖的miRNA基因转录靶向TGF-β/Smad3信号通路可为肾纤维化的治疗提供新的策略。
研究发现,TGF-β超家族中的新型分子lefty-1能够抑制TGF-β信号通路的活化,减少上皮间质转化;过表达lefty-1可明显抑制TGF-β1/Smad信号通路的活化,减少上皮间质转化及ECM的合成,减轻肾脏间质纤维化[40]。在糖尿病诱导的肾纤维化小鼠中,纤维生长因子21能够抑制TGF-β/Smad2/3的激活,从而抑制Smad2/3向细胞核转移,减少肾上皮间质转化,减轻糖尿病诱导的肾纤维化;进一步研究发现,敲除Smad3可减少Ⅰ型胶原及Ⅳ型胶原蛋白的沉积,抑制肾纤维化的进展[41-42]。
非经典的TGF-β信号通路也参与了肾纤维化的发生。在阻塞性肾病小鼠的肾组织中,TGF-β1表达上调,激活下游的Wnt/β-连环蛋白(β-catenin)信号通路,增加Ⅰ型胶原及纤连蛋白的沉积[42]。五倍子酸盐能够抑制肾细胞中TGF-β1/糖原合成酶激酶-3β(glycogen synthase kinase-3β,GSK-3β)/β-catenin信号通路,从而抑制TGF-β1诱导的上皮间质转化,减轻肾纤维化[43]。此外,灵芝酸能够抑制TGF-β/Smad及MAPK信号通路,从而抑制上皮间质转化及纤连蛋白的表达,减少ECM的沉积,抑制肾纤维化进程[44]。
2.5 骨髓纤维化 骨髓纤维化是由于促纤维生长因子过度释放,骨髓造血组织被结缔组织取代所引起的骨髓增殖性疾病,包括原发性骨髓纤维化和继发性骨髓纤维化,其病理特征主要为骨髓纤维化、贫血、血管增生、肝脾肿大及ECM增多[45]。研究发现,促炎细胞因子TGF-β在骨髓纤维化的发生中起重要作用,在促进骨髓纤维化及骨髓增生中起双重作用[46]。在原发性骨髓纤维化患者的骨髓组织中,可检测到骨髓纤维化发生前期TGF-β1的表达增加,从而诱导血管生成及纤维化的发生,且TGF-β1的表达与微血管密度、纤维化程度呈正相关[47]。
研究发现,在骨髓纤维化发生过程中,TβR-Ⅰ、 TβR-Ⅱ表达水平降低,Smad1、Smad2及Smad4的表达减少,经典的TGF-β/Smads信号通路失活,非经典的TGF-β信号通路参与了骨髓纤维化的发生[48]。 在骨髓纤维化患者的骨髓及脾脏中,非经典的TGF-β1信号通路活化,激活下游的Hedgehog信号及p53信号,从而导致成骨细胞的分化及造血功能失调[49]。 另有研究发现,在原发性骨髓纤维化CD34+细胞中,TGF-β1上调下游分子miR-382-5p的表达,降低超氧化物歧化酶2(superoxide dismutase 2,SOD2)的活性,导致活性氧簇(reactive oxygen species,ROS)的积聚,引起氧化应激及炎症反应;抑制TGF-β1/miR-382-5p/SOD2的信号转导可恢复SOD2的活性,减少ROS的生成,减轻骨髓纤维化[50]。
2.6 胰腺纤维化 胰腺纤维化是一种消化系统疾病,是慢性胰腺炎的典型病理表现;其病理机制是胰腺星状细胞活化,ECM合成增加、降解减少导致了ECM聚积[51]。已有研究发现,TGF-β1是胰腺纤维化发生过程中的主要调控因子,TGF-β信号转导参与了胰腺相关疾病的调控,在细胞凋亡、炎症反应及致癌作用等方面发挥重要作用[52]。
在大鼠胰腺星状细胞中,TGF-β/Smad2/3信号通路参与了胰腺纤维化的形成,而香豆素二甲醚能够抑制TGF-β/Smad2/3信号通路的活化,上调Smad7的表达,从而抑制星状细胞的增殖及上皮间质转化,减轻氧化应激反应及纤维化的进展[53]。进一步研究发现,慢性胰腺炎小鼠胰腺组织中TGF-β的分泌增加;在胰腺星状细胞中,TGF-β激活下游的Smad2/3,上调胶原蛋白、纤连蛋白1及α-肌动蛋白的表达;胡椒碱能够抑制TGF-β/Smad2/3信号通路的活化,降低纤维调控因子的表达,改善胰腺纤维化[54]。此外,TGF-β1也能激活下游的RhoA/Rock信号通路,增加Ⅰ型胶原蛋白及纤维状肌动蛋白的表达,诱导胰腺纤维化;而内皮抑制素能够抑制TGF-β1/RhoA/Rock信号通路的活化,从而抑制TGF-β1诱导的胰腺纤维化[55]。由此可知,经典的TGF-β/Smad信号通路和非经典的TGF-β信号通路均可参与胰腺纤维化的发生。
综上所述,TGF-β信号通路参与了肺、肝、心肌、肾脏及胰腺等纤维化疾病的发生,抑制经典的TGF-β/Smads信号通路及非经典的TGF-β信号通路的信号传递,对纤维化疾病具有潜在的治疗作用。由于不同器官在结构及功能上的差异,TGF-β信号通路对各纤维化疾病的调控机制也不完全相同,但仍有相似之处,如调控纤维化相关细胞的活化、胶原的合成及ECM的聚积。临床上针对TGF-β及其上游信号分子的药物研发为纤维化疾病的治疗带来了希望,已研发的靶向治疗药物包括吡非尼酮(pirfenidone)、尼达尼布(nintedanib)、TGF-β单克隆抗体(FG-3019、LY2382770)、干扰素-γ及维A酸等。由于TGF-β生物活性高且应用广泛,已有的药物很难对其进行单一调控,且对其他脏器的不良反应较大。目前,靶向经典的TGF-β信号通路下游的信号分子Smad3、Smad7及非经典的TGF-β信号通路成为新的研究方向,而筛选出纤维化发生相关的靶点,可为纤维化疾病治疗药物的研发提供理论 支持。
猜你喜欢
中老年保健(2022年2期)2022-11-25 23:46:31
昆明医科大学学报(2022年4期)2022-05-23 13:04:50
昆明医科大学学报(2021年8期)2021-08-13 08:59:50
云南医药(2021年3期)2021-07-21 05:40:30
国际呼吸杂志(2019年21期)2019-11-25 09:52:20
Coco薇(2017年12期)2018-01-03 21:27:09
天然产物研究与开发(2016年6期)2016-06-05 10:29:30
现代食品(2016年14期)2016-04-28 08:10:07
广东海洋大学学报(2015年4期)2016-01-13 08:39:40
中国现代医学杂志(2015年26期)2015-12-23 11:04:22
