
基因检测确诊纤毛结构正常的原发性纤毛运动障碍2例病例报告
2020-12-16 03:06付文龙田代印范京川代继宏
中国循证儿科杂志 2020年5期
李 莹 付文龙 田代印 耿 刚 范京川 代继宏
1 病例资料
例1:女,9月龄,因“慢性湿性咳嗽6个月”入重庆医科大学附属儿童医院(我院)治疗。3月龄起开始出现反复咳嗽伴痰鸣音,伴流涕和鼻塞,无喘息、气促和呼吸困难,偶有发热。外院多次诊断为“支气管炎”、“肺炎”等,予抗感染治疗1周左右,咳嗽、咳痰缓解。患儿为G1P1,胎龄30+5周,生后有新生儿呼吸窘迫,予呼吸机辅助通气,具体治疗不详。生长发育史正常。入院查体双肺呼吸音对称,可闻及少许中粗湿啰音。血常规、肝肾功能和免疫功能检查未见异常。心脏彩超提示镜面右位心,主动脉右弓右降。腹部B超提示胃左位,肝右位。胸部CT显示肺炎,左肺3叶、右肺2叶(图1A)。
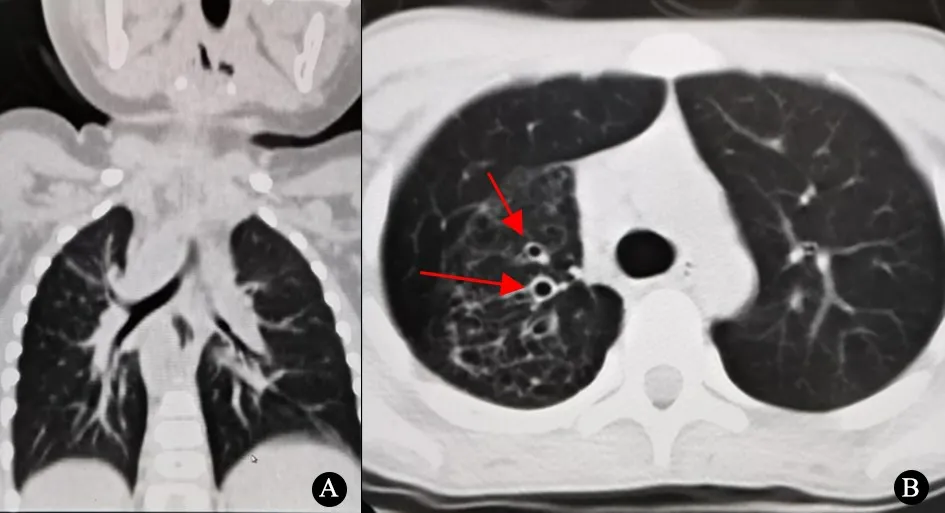
图1 2例患儿的CT表现
例2:男,14岁,因“反复咳嗽、咳痰2年余,加重3个月”入我院。 2年前开始出现持续性咳嗽伴咳脓痰,伴鼻塞、流涕,外院诊断“慢性鼻窦炎”。自幼有双耳流脓(起病具体时间不详),曾诊断“化脓性中耳炎伴鼓膜穿孔”。咳嗽、咳痰平均每1~3个月急性发作1次,每次抗感染、化痰治疗1~2周可缓解。患儿为G2P2,足月顺产,新生儿期无住院病史,生长发育史正常。患儿的姐姐身体健康。否认类似支气管扩张家族史,否认3代以内有近亲婚配。入院查体双侧胸廓对称,双肺呼吸音对称、闻及少许粗湿啰音。血常规、肝肾功能、免疫功能检查未见异常,结核干扰素试验阴性。胸部CT提示弥漫性支气管扩张(图1B)。
2例患儿均行支气管镜下支气管黏膜活检,在透射电镜(TEM)下分析纤毛结构,均提示呼吸道纤毛“9+2”轴突超微结构正常(图2)。
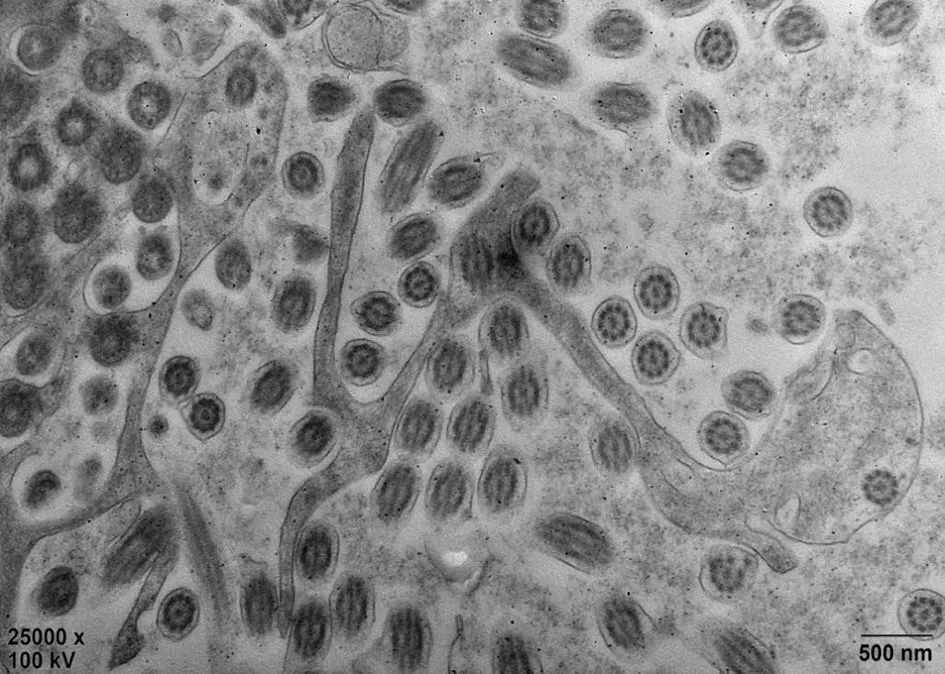
图2 电子显微镜显示例1患儿纤毛正常超微结构
2 基因检测
2例患儿临床特征典型,高度疑诊原发性纤毛运动障碍(PCD,MIM# 242650),为进一步明确诊断,经家长知情同意后,分别抽取患儿及其父母外周血2 mL,乙二胺四乙酸(EDTA)抗凝,行基因测序(北京迈基诺基因检测公司),例1行全外显子基因测序,例2行呼吸系统疾病基因包测序(包括804个相关基因)。通过Sanger测序进行验证并分析父母来源。
例1检测到DNAH11基因(NM_00127715)49号外显子c.7999C>T(p.Q2667X)无义突变,65号外显子c.10691+2T>G剪接突变,分别来自患儿母亲和父亲。例2检测到HYDIN基因(NM_001270974)84号外显子c.14641delG (p.V4881fs)移码突变,46号外显子c.7159-1G>A剪接突变,分别来自患儿母亲和父亲。
根据美国医学遗传学与基因组学学会(ACMG)联合美国分子病理学会(AMP)2015 年制订的“基因序列变异的解释标准和指南”(简称指南)行致病性分析。例1的2个突变位点和例2的2个突变位点均满足指南中PVS+PM2+PM3+PP4 4个致病支持性证据,判定为致病性突变,支持性证据如下。①PVS:剪接突变、无义突变和移码突变均为零效变异,可能导致基因功能丧失。②PM2:HGMD 数据库无这4个位点的相关报道。生物信息学蛋白功能预测软件 SIFT、PolyPhen_2、REVEL预测均为未知。在千人基因组、ESP6500、gnomAD和ExAC 4个正常人群数据库中未检索到这些位点或提示正常人不携带。③PM3:该病为隐性遗传病,与另1个致病变异反式存在(与另1个致病突变组成复合杂合)。④PP4:患儿的临床表现与PCD表型高度吻合。
3 讨论
PCD是一种罕见的常染色体隐性遗传病,发病率1/15 000~1/20 000[1],其特征为纤毛运动功能受损导致黏液纤毛清除无效。婴幼儿时期高度提示PCD的临床表现包括:新生儿呼吸窘迫、反复上呼吸道感染(慢性鼻-鼻窦炎、慢性中耳炎)、早期出现的反复下呼吸道感染和内脏转位等[2, 3]。TEM下呼吸道纤毛结构异常是诊断PCD的传统标准。然而,文献报道,>30%的PCD患者纤毛超微结构正常[4-6]。因此,TEM提示纤毛轴突超微结构正常不能完全排除PCD,需结合致病基因检测等其他检测方法进行诊断。理论上,任何涉及纤毛装配或功能的基因突变都可能导致PCD,目前已知40余个相关基因,大多为常染色体隐性遗传,也有部分为X连锁隐性遗传[7]。本文2例临床高度疑诊PCD的患儿TEM下纤毛结构正常,基因检测分别发现DNAH11和HYDIN基因发生复合杂合突变,均为致病性突变,从而确诊PCD。
DNAH11基因编码一种外动力蛋白臂(ODA)重链蛋白,其突变并不影响其他外动力蛋白(DNAH5、DNAH9)在纤毛轴丝上的位置[8],但引起纤毛摆动异常,表现为异常刚性搏动模式、纤毛弯曲能力减弱、摆动过度,TEM和免疫组化则显示,呼吸纤毛轴突超微结构正常,外动力蛋白臂完整[9]。既往研究对TEM下观察发现纤毛结构异常的PCD患者行基因检测,均未发现DNAH11基因突变[10]。关于DNAH11基因突变在PCD患者中的比例,土耳其报告为8.7%[11],中国约为14.3%[6]。在纤毛轴突超微结构正常的PCD患者中,DNAH11基因突变约占22%[10]。目前关于PCD基因型及临床表型关系的研究尚不多,Schwabe等[9]在既往的一项家系研究中描述DNAH11基因突变所致的PCD患者的临床症状包括慢性耳道感染、反复的鼻部症状及慢性肺部感染、新生儿呼吸窘迫、全内脏转位(Kartagener 综合征)等,与本文患儿的临床表现基本一致。
HYDIN最初被认为是一种导致脑积水的基因,编码大脑中央装置纤毛蛋白,该基因突变导致的脑积水患者在常规TEM下观察大脑中央装置无明显结构异常[12]。后续研究发现HYDIN基因突变还可引起PCD,患者表现为中耳炎、鼻窦炎、肺炎等反复感染症状,在TEM下观察运动纤毛超微结构完全正常[13]。既往研究报道,德国和法罗群岛人群HYDIN突变在所有PCD患者中<1%[13],土耳其人群中则为10.9%[11]。推测HYDIN基因突变在不同人群PCD患者中的比例差异较大。本文伴有HYDIN突变的例2符合上述临床特征,但12岁出现首发症状,与PCD自幼起病的特点不符合。可能的原因包括:①患儿及家长对其既往病情存在回忆偏倚。②PCD临床异质性较大,有文献报道首发症状可出现在1月龄至11岁[11]。由于HYDIN编码呼吸纤毛的中心复合体,不影响动力蛋白的功能,因此患儿胚胎发育时期结纤毛功能不受影响,故认为该基因突变不会引起内脏转位[13]。
此外,既往研究发现,一些编码轴丝辐射状头蛋白的基因发生突变也可导致纤毛结构正常的PCD。例如,RSPH9突变患者其纤毛超微结构完全正常[14]。RSPH4A突变者约50%具有正常的纤毛超微结构[15]。CCDC164和GAS8均为编码连接蛋白和动力蛋白调节复合物(N-DRC)的基因, TEM分析发现这2种基因突变所致的PCD纤毛超微结构正常[16, 17]。一种由RPGR基因突变导致的伴有视网膜变性的PCD综合征,患儿也可以具有正常的纤毛超微结构[18]。
目前尚无单一诊断方法可作为确诊PCD的金标准,使得临床确诊PCD仍有一定困难。对于具有典型临床表现但纤毛超微结构没有明显缺陷的PCD患儿,其诊断仍具有挑战性。PCD患儿虽然存在较强的异质性,但临床上早期起病的慢性咳嗽,反复上、下呼吸道感染,内脏转位及新生儿期不明原因呼吸窘迫,均对PCD 的诊断有高度提示作用[19]。同时,研究表明,纤毛超微结构正常的PCD患儿的临床特征与伴有动力臂缺失等纤毛结构异常的患者相比基本无差异[4]。 因此,临床特征仍是许多患者诊断PCD的首要和重要线索。
本文的不足之处在于,由于检测条件限制,未使用高速扫描电镜评估纤毛运动功能。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
自然杂志(2022年3期)2022-08-18
农业工程学报(2022年4期)2022-04-24
医学研究生学报(2021年4期)2021-12-02
临床与实验病理学杂志(2021年3期)2021-04-25
中国生殖健康(2020年4期)2021-01-18
中华皮肤科杂志(2019年5期)2019-06-24
中国生殖健康(2018年4期)2018-11-06
天津农业科学(2015年12期)2015-12-03
军事体育学报(2015年2期)2015-02-27
