
以铂基高指数晶面纳米晶为载体负载过渡金属催化甲醇电氧化
2020-12-16 06:39王翊博李昀芮谢枫苡徐展硕
石油化工 2020年11期
王翊博,李昀芮,谢枫苡,徐展硕,王 耀,张 鑫
(中国石油大学(北京) 重质油国家重点实验室 化学工程与环境学院,北京 102249)
生物质燃料(如甲醇、乙醇、乙二醇)的高效氧化可以应用于质子交换膜燃料电池[1]。在燃料电池应用中,过渡金属尤其是贵金属Pt及其复合纳米结构受到广泛关注,并在电催化反应中表现出极其优异的性能,很难完全被其他材料取代。但至今质子交换膜燃料电池的规模化应用仍难以实现,其中一个主要原因是电极催化剂中贵金属Pt的价格及使用量过高[2]。此外,从应用性能看,催化剂的Pt利用率、本征催化活性、抗毒性能及电化学稳定性均有待进一步提高。这些问题的存在极大限制了质子交换膜燃料电池技术的广泛应用。
贵金属纳米晶的电催化性能与它的形貌和组成密切相关,高指数晶面(HIF)是Miller指数均大于1的晶面。HIF具有较高的表面自由能和较低的配位数,因此具有良好的催化活性,但是较高的表面自由能和丰富的不饱和原子导致它的表面容易发生结构重排。在反应过程中,HIF的结构趋于消失,并不稳定[3]。因此,为了提高裸露HIF的Pt基合金纳米晶的稳定性,将Pt与其他非贵金属复合,设计Pt基双元、甚至多元合金纳米结构是改善Pt基催化剂性能的有效方法[4]。当非Pt金属作为助催化剂/共催化剂存在时,能够通过金属组分间几何/电子结构的相互作用所引起的集团效应、配体效应、晶格应力效应及协同效应等对Pt的分散状态、表面电子结构及相应催化性能进行调变[5],从而可以稳固HIF的表面结构,提高它的甲醇电氧化反应性能[6]。
在目前的研究中,已有将非贵金属或非金属作为活性助剂植入与HIF结合的Pt基纳米晶的表面以增强HIF稳定性的方法[7],在获得较高的甲醇、乙二醇氧化反应活性方面也有不错的效果[8]。植入晶体表面的活性助剂可以改变/调节微晶表面的物理化学性质(组成和结构)[9],赋予Pt基纳米晶以优异的催化活性。目前研究较多的是将Au,Rh,Mo,Fe等金属作为活性助剂植入纳米晶的方法,本课题组通过在合成Pt3Mn凹面立方体(简称Pt3Mn)前体的过程中引入Mo金属前体,提高了纳米晶的稳定性和催化活性,且促进了COHx向CO2的转化。金属Sn基材料具有较好的电化学性能,廉价且无毒副作用,目前报道的一些Sn基纳米结构材料具有优异的电化学性能及循环稳定性[10]。因此本课题组尝试将Sn作为活性助剂植入到Pt3Mn的HIF。
本工作以Pt3Mn为载体,引入非贵金属Sn,制备了Sn/Pt3Mn催化剂,对催化剂进行了XRD和TEM表征,并将其用于甲醇电氧化反应,探讨了催化剂的构效关系。
1 实验部分
1.1 催化剂的制备
采用两步法合成Sn/Pt3Mn催化剂[1]。首先称取0.220 g聚乙烯吡咯烷酮和0.088 g甘氨酸加入到内衬聚四氟乙烯的不锈钢高压反应釜中,量取5 mL 100 mmol/L的H2PtCl6溶液和4 mL 1.66 mmol/L的MnCl2溶液置于高压反应釜中,得到黄色混合物;将上述混合物先进行磁力搅拌,再进行超声搅拌,形成均匀的黄色混合液体,然后密封反应釜,将其置于烘箱中,在200 ℃下反应6 h,制得Pt3Mn;随后分别加入 2,9,45 μL 的 20 mmol/L SnCl4溶液,继续在200 ℃下反应2 h以合成Sn含量(质量分数,下同)为0.1%,0.5%,2.4%的Sn/Pt3Mn催化剂,分别记为0.1%Sn/Pt3Mn,0.5%Sn/Pt3Mn,2.4%Sn/Pt3Mn。将反应釜中合成的试样用水-乙醇混合溶液进行离心洗涤,并在-50 ℃下冷冻干燥待用。
1.2 催化剂的活性及稳定性测试
采用上海辰华仪器有限公司CHI 760E电化学工作站的三电极系统测量Sn/Pt3Mn催化剂对甲醇电氧化的催化活性及稳定性,以饱和甘汞电极为参比电极、铂丝电极为对电极、玻碳电极为工作电极。先用氮气清洗三电极系统30 min,连接电极,开启电化学分析仪,测量Sn/Pt3Mn催化剂的活性及稳定性。
在测试前,将催化剂在紫外线灯(10 W,发射波长185 nm和254 nm的光)下处理12 h,以除去封端剂。用氧化铝粉处理玻碳电极后,将6 μL的催化剂墨水滴到玻碳电极上,然后将2 μL的0.05%(w)萘酚乙醇溶液滴加到催化剂上。在室温下,以50 mV/s的扫描速率在饱和N2、0.5 mol/L H2SO4和2 mol/L甲醇混合溶液中进行循环伏安法(CV)扫描,以进行电极表面的清洁和催化剂的活化。
根据氢脱附峰面积计算电化学活性面积,再计算得到电流密度,用电流密度表示催化剂的电氧化面积比活性,电位范围为0.04~1.00 V(可逆氢电极(RHE))。通过计时电流法在0.7 V(RHE)下测试催化剂的稳定性。
1.3 催化剂的表征
采用日本电子株式会社JEM-2100型透射电子显微镜进行TEM表征,并采用同公司的Tecnai G2 F20 S-Twin型高分辨率透射电子显微镜进行HRTEM表征。采用Bruker公司的D8 Advance型X射线衍射仪进行XRD表征,CuKα辐射(波长0.154 06 nm),管电压40 kV,管电流40 mA,扫描速率 5 (°)/min,步长0.02°。
2 结果与讨论
2.1 催化剂的表征结果
2.1.1 TEM和HRTEM表征结果
Pt3Mn和Sn/Pt3Mn试样的TEM和HRTEM照片见图1。由图1(a)可见,Pt3Mn的平均尺寸为(48.9±15) nm(对角线顶点到顶点);由图1(e)可见,晶格条纹的间距为0.203 nm,略小于 Pt 的面心立方结构(fcc)的(200)晶面的晶格间距,说明Pt与Mn是以合金态存在的,揭示了Pt-Mn合金的形成。从图(b)~(d)可看出,Sn/Pt3Mn显示出完美的凹形结构,这种凹形结构证实成功合成了HIF纳米晶。

图1 Pt3Mn和Sn/Pt3Mn试样的TEM和HRTEM照片Fig.1 TEM and HRTEM images of Pt3Mn and Sn/Pt3Mn samples.
在Pt3Mn中引入Sn的前体,通过原位外延生长制备了Sn/Pt3Mn催化剂。Pt3Mn的形成是晶体的直接生长过程,因此Sn/Pt3Mn纳米晶体的结构将优先沿着Pt3Mn衬底的晶体方向生长,掺杂Sn后,凹面的形态保持不变。值得注意的是,在0.1%Sn/Pt3Mn,0.5%Sn/Pt3Mn,2.4%Sn/Pt3Mn试样中发现有三元合金壳,壳宽分别为2.93,3.67,5.55 nm,且壳的宽度随Sn含量的增加而增大,这表明Pt3Mn的表面可能由于Sn的插入而重构。此外,在引入Sn后,Pt富集在合金表面,伴随着HIF的再生,导致催化剂的电催化活性提高。
TEM和HRTEM照片从微观尺度证明加入Sn并未改变Pt3Mn的形貌,并形成了HIF的Pt-Mn凹面立方体合金。
2.1.2 XRD表征结果
Pt3Mn和Sn/Pt3Mn试样的XRD谱图见图2。
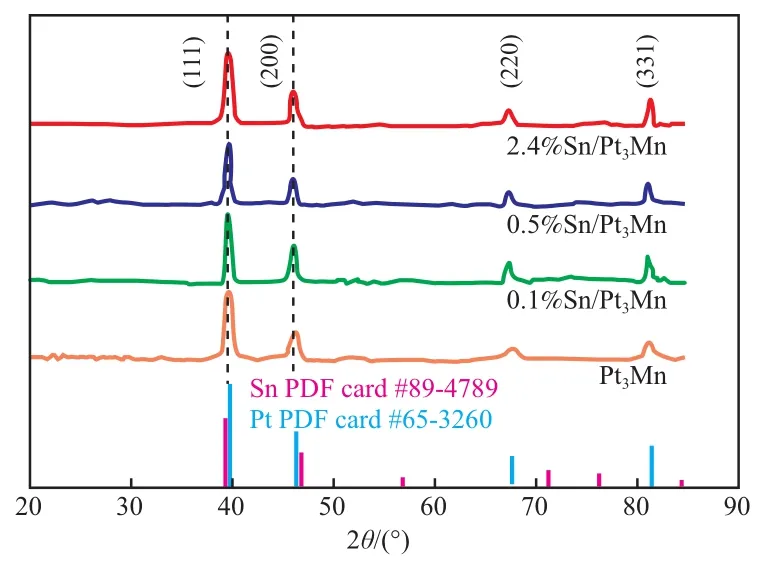
图2 Pt3Mn和Sn/Pt3Mn试样的XRD谱图Fig.2 XRD spectra of Pt3Mn and Sn/Pt3Mn samples.
由图2可见,四种试样中的XRD谱图显示了类似Pt的fcc结构。对比Sn/Pt3Mn与纯Pt、纯Sn试样的XRD谱图可看出,Pt-Mn合金的(200)峰相比纯Pt略向左移,而(111)峰则略向右移,这说明已将Sn成功引入到Pt-Mn合金中。本工作采用水热法引入Sn,并形成HIF。在XRD谱图中没有Sn或其氧化物的衍射峰,这进一步证明Sn是以原子的形式嵌入在Pt-Mn合金中。值得注意的是,Pt3Mn和Sn/Pt3Mn试样的晶面取向均为(111)和(200),表明Sn的引入几乎不会改变Pt3Mn纳米晶体的晶面取向。
2.2 催化剂的甲醇电催化性能
为了探索复合材料中Sn的作用以及三组分之间的协同作用对电催化性能的影响,对Sn/Pt3Mn催化剂的电化学性能进行测试,并以商业 Pt/C催化剂作为对比。在测试前,将所有的催化剂置于 0.5 mol/L H2SO4和2 mol/L甲醇的电解液中绘制CV曲线(见图3),目的是活化催化剂。以下所有的电流密度均以Pt在0.5 mol/L H2SO4和2 mol/L甲醇电解液中扫描得到的CV曲线中的氢脱附峰面积为计算标准。
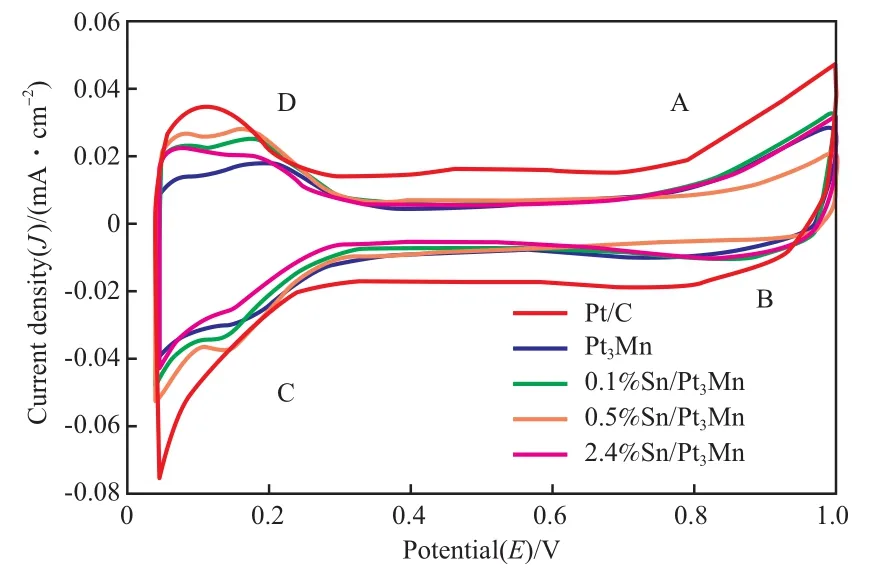
图3 不同催化剂的CV曲线Fig.3 Cyclic voltammetry(CV) curves of different catalysts.
由图3可知,Sn/Pt3Mn催化剂具有与Pt/C催化剂相似的氧化还原活性,Pt氧化物的形成区(区域A)和还原区(区域B),以及氢的吸收区(区域C)和解吸区(区域D)的形状、走势均相同,说明Sn/Pt3Mn催化剂与Pt/C催化剂有着相似的氧化还原行为,即它们的氧化还原活性相似。
将区域D的面积积分得到Pt的氢脱附峰面积,以此为基础计算得到电流密度,用电流密度表示催化剂的电氧化面积比活性,结果如图4所示。
0.1%Sn/Pt3Mn,2.4%Sn/Pt3Mn,0.5%Sn/Pt3Mn催化剂对甲醇的电氧化面积比活性分别为4.43,5.09,8.01 mA/cm2, 是 Pt3Mn催 化 剂(2.82 mA/cm2)的1.57,1.81,2.84倍,是商用Pt/C催化剂(2.74 mA/cm2)的1.62,1.86,2.92倍。这表明经Sn修饰后,Sn/Pt3Mn催化剂对甲醇的电催化氧化性能得到极大的提高。但Sn含量过高反而会使催化剂的性能减弱,这可能是由于过多的Sn覆盖了表面的Pt原子,减少了活性中心的表面密度。催化性能增强可能是由Pt与其他3d过渡金属之间的界面极化引起的,3d过渡金属的功函数会导致Pt表面的电子富集,从而促进电催化过程[11]。
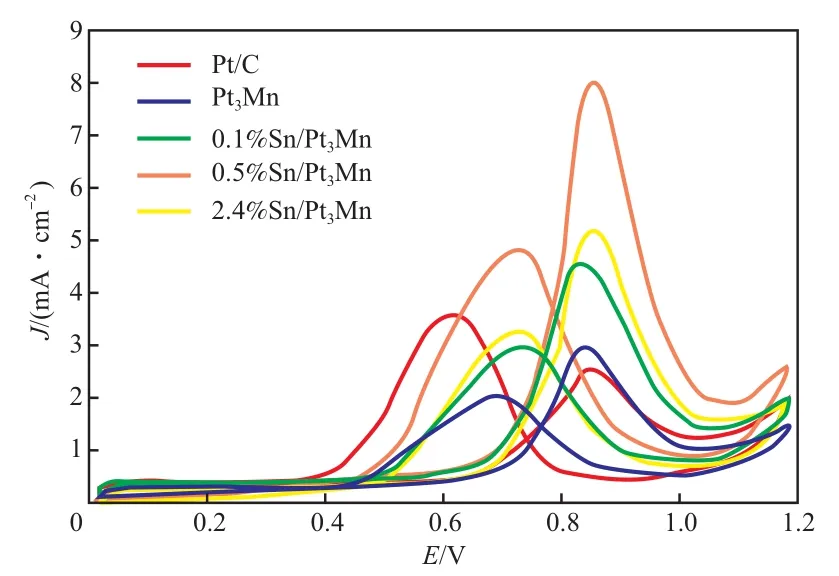
图4 Sn/Pt3Mn,Pt/C,Pt3Mn催化剂的电氧化面积比活性Fig.4 Sn/Pt3Mn,Pt/C,Pt3Mn electro-oxidation area-specific activity diagram.
为了能够在更接近于真实甲醇燃料电池的实际工作条件下评价催化剂的电化学稳定性,选取甲醇燃料电池的典型工作电位0.7 V(RHE)作为测试电位,得到不同催化剂的计时电流曲线,结果如图5所示。
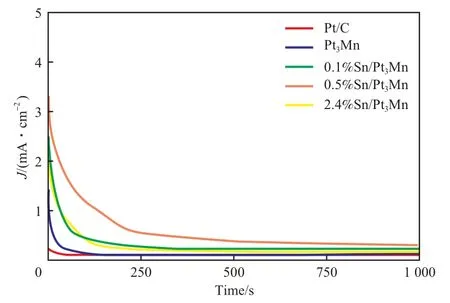
图5 不同催化剂的CV扫描计时电流曲线Fig.5 CV sweep chronoamperometric curves of different catalysts.
从图5可看出,在250 s前三个催化剂的电流密度都出现了明显的下降,随着时间的延长,电流的下降幅度相近;500 s后,三个催化剂的电流密度均趋于稳定。从电流密度下降的趋势和最后稳定时的电流密度可以发现,0.1%Sn/Pt3Mn,0.5%Sn/Pt3Mn,2.4%Sn/Pt3Mn催化剂均表现出比Pt3Mn及商用Pt/C催化剂更优异的电化学稳定性。这进一步证明了Sn修饰的Pt3Mn催化剂,尤其是0.5%Sn/Pt3Mn催化剂在燃料电池中具有良好的应用前景。
0.5%Sn/Pt3Mn催化剂可以在施加的电势下保持初始结构的原因可能是由于Sn的氧化。本课题组通过在改性Pt3Mn前体上引入Mo组分,提高了纳米晶的稳定性和催化活性,且提高了COHx向CO2的转化。我们推测主要是位于催化剂表面附近的Sn在电化学处理中优先被氧化,因为它的氧化电势为0.10 V,而它的RHE低于Mn(1.224 V)和 Pt(1.180 V)[12],它优先进行连续的氧化/还原过程,因此可保护Pt3Mn/Sn催化剂的最外层。Cao等[13]也证明了Sn的掺杂可以降低表面3d过渡金属的平衡浓度、并增加表面层中Pt/Sn空隙生成焓,这限制了过渡金属的溶解速度。以上分析结果表明,稳定性实验后Pt3Mn的活性变差是由于Mn的溶解和HIFs结构被破坏,而Sn/Pt3Mn的形态在电化学测量后仍保持完整。从Pt3Mn和0.5%Sn/Pt3Mn催化剂的元素组成变化可以推断出,Sn的掺入可以稳定Mn原子、并防止Pt迁移[14],最终使催化剂可以保持优异的催化活性[15]。综上所述,Sn掺入后并不改变Pt/Mn合金的形貌,但通过降低表面3d过渡金属的平衡浓度、增加空隙生成焓的方式限制过渡金属的溶解速度,稳定了HIF结构,使催化剂保持优异的活性。
根据以上结果,提出如图6所示的Sn/Pt3Mn结构。这种独特的结构具有以下特性:1)表面看,Pt3Mn是Sn的“载体”,事实上Sn组分被注入到Pt3Mn外层的表面晶格中。2)XRD和TEM表征结果显示,Pt,Mn,Sn元素均匀分布在Sn/Pt3Mn凹面立方体的壳体中,并形成Pt-Mn-Sn三元合金;而Pt3Mn中Pt和Mn元素则为二元合金,Mn分布均匀,并在凹面立方体的内芯中形成Pt-Mn二元合金。3)添加少量的Sn导致壳中Pt 4f的结合能下降,而Pt3Mn二元合金的内核中Pt 4f的电子效应没有改变。4)在Sn/Pt3Mn催化剂中,形成了相对较强的Sn—Mn键,而不是Pt—Mn键和Pt—Mn键,导致Mn在酸溶液中的反溶解。
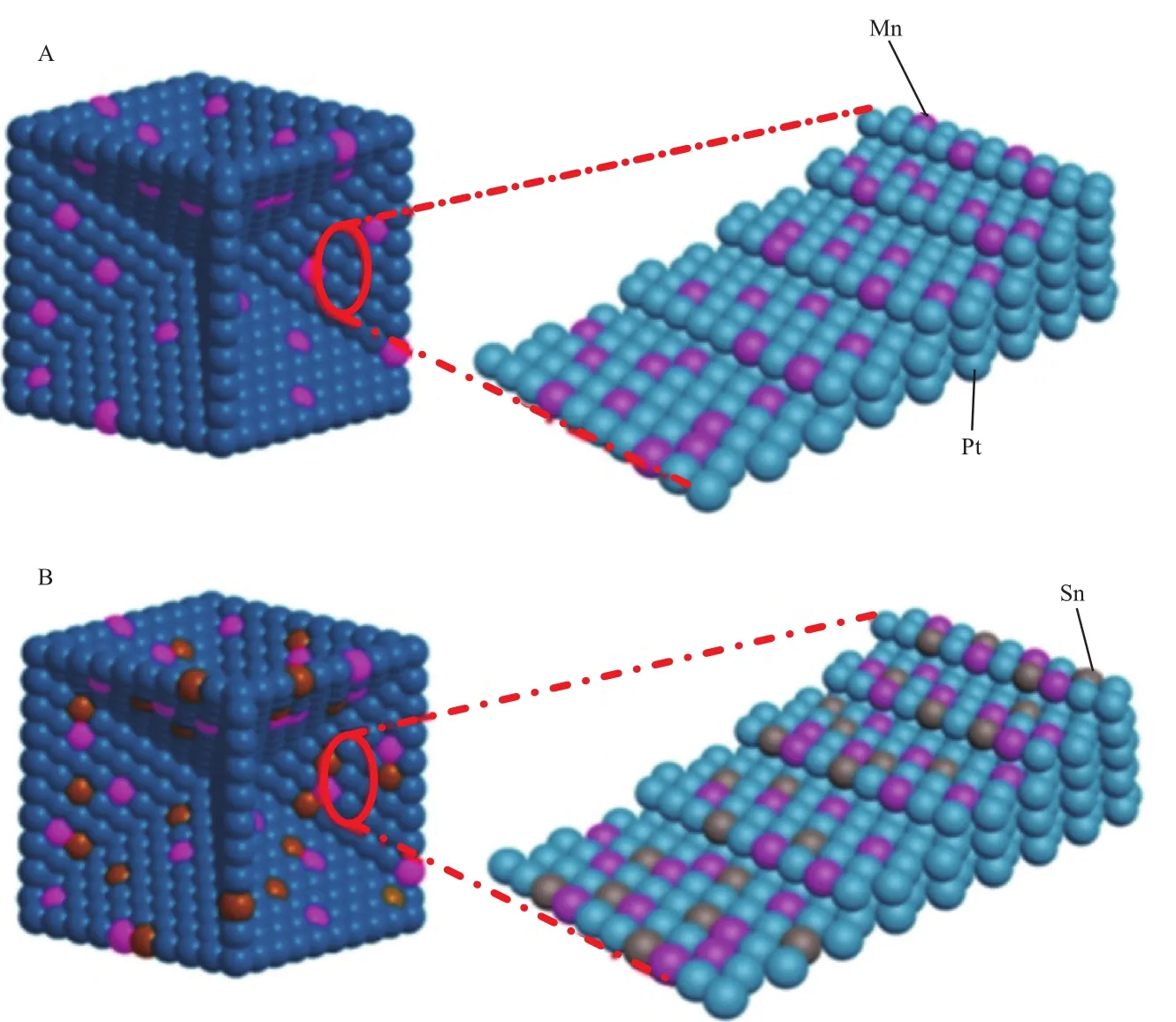
图6 Pt3Mn(A)和Sn/Pt3Mn(B)催化剂表面的示意图[1]Fig.6 Schematic diagram of the surface of Pt3Mn(A) and Sn/Pt3Mn(B) catalyst[1].
从催化性能上来说,Sn修饰的Pt基HIF纳米晶结构与不加入Sn的Pt基HIF纳米晶结构相比,活性和稳定性均明显提高,且随着Sn含量的增加,活性呈现火山形变化趋势,即随着Sn含量的增加,催化剂的活性和稳定性均先增大再减小,其中0.5%Sn/Pt3Mn催化剂的活性和稳定性最佳。Sn作为“牺牲”金属,优先进行氧化还原反应,保护了作为催化活性位点的Pt-Mn金属,提高了催化剂的稳定性。其次引入的Sn降低了Pt、Mn表面3d过渡金属的平衡浓度,并增加了表面层中Pt/Sn空隙生成焓,这限制了Pt-Mn金属的溶解速度,进一步增强了催化剂的稳定性和催化活性。
3 结论
1)通过将非贵金属作为活性助剂注入结合有HIF的Pt纳米晶体的表面,可设计稳定的新型结构催化剂,从而赋予Pt催化剂优异的稳定性和活性。
2)以Pt3Mn为载体制备的Sn/Pt3Mn催化剂保持了Pt3Mn的形态,但催化活性和稳定性提高,其中,0.5%Sn/Pt3Mn催化剂显示出优异的甲醇电催化性能,面积比活性是Pt3Mn的2.84倍。
猜你喜欢
化工管理(2022年14期)2022-12-02
铝加工(2022年3期)2022-11-24
材料与冶金学报(2022年2期)2022-08-10
中国化肥信息(2022年3期)2022-05-05
汽车实用技术(2022年4期)2022-03-07
口腔护理用品工业(2021年4期)2021-11-02
安徽工业大学学报(自然科学版)(2021年3期)2021-09-08
粉末冶金技术(2021年3期)2021-07-28
装备维修技术(2020年5期)2020-11-20
分析化学(2018年1期)2018-01-18
