
基于Abraham模型估算有机物表面张力
2020-12-16 06:40左丽华陈六平
石油化工 2020年11期
左丽华,陈六平
(1. 东华理工大学 化学生物与材料科学学院,江西 南昌 330013;2. 中山大学 化学学院,广东 广州 510275)
纯液体表面张力(σ)是物理和传递过程中重要的基础数据,在化工生产、工程设计等方面应用广泛。σ的测试方法很多,至今文献已公布的数据也较多,Jasper[1]曾系统整理了 1847—1969年间所公布的2 200多种物质在不同温度下的σ;Vargaftik[2]也提供了不少不同温度下σ的实验数据。随着精细化工的快速发展,现有温度范围内的σ数据已无法满足应用需求。而每年新递增的有机化合物种类繁多,逐一测定的实验量太大,因此发展纯液体尤其是纯有机物σ的计算方法有着重要的理论和实用意义。σ的估算方法很多,其中应用较为广泛、且计算精度在可接受范围内的估算纯物质σ的方法有基团贡献法[3-5]、人工神经网络法[6-7]、基于构效关系预测法[8-11]及其他估算方法[12-14]。这些估算方法大多仅限于结构简单的物质及有限的温度范围,且计算繁复、误差较大,有完善的空间。
本工作提出一种将σ这一宏观物性与微观分子间作用力相关联的一种新的计算方法。根据线性溶剂化能量关系(LSERs)理论和经验方程,提出了在不同温度下估算纯有机物σ的关联式。利用所得关联式对2 628个纯组分体系中的10个同系物进行了回归,得到了σ的关联方程,并对方程的相关性和外推适用性进行考察。
1 方程的推导
前期曾尝试从物质的本质出发,在分子间相互作用的水平上,结合LSERs理论[15],采用Abraham分子结构参数,建立一个新的LSERs能量模型[16],见式(1);并结合温度的影响,对纯物质的蒸发焓和物质相互扩散系数进行估算,取得了良好的预测精度[17-18]。

式中,SP为系统性质;C为常数;e为过摩尔折射率系数;E为过摩尔折射率,cm3/mol;j为极化率系数;J为极化率,C·m2/V;a为氢键酸度系数;A为氢键酸度;b为氢键碱度系数;B为氢键碱度;v为特征分子体积系数;Vx为Abraham和Mc Gowan特征分子体积(反映分子大小),10-2cm3/mol。
在此基础上,利用上述Abraham特征线性溶剂化能量模型,对10类同系物的σ数据进行拟合,提出估算纯有机物σ的计算模型,拟为纯有机物σ的计算提供一种可供选择的方法。
由热力学原理及σ的定义可知,如果要将一个分子从液体内部移到表面,就需要克服液体内部周围分子对它的吸引力而做功。在恒温恒压可逆的条件下,形成新表面所消耗的功,将转化为表面层分子比内部分子多余的Gibbs自由能,即表面自由能。若体系组成不变,则可逆地使表面积增加(dAs)所需的功(W)为 δW=σdAs,又 -δW= d(G)Tp,得式(2)。

式中,σ为纯液体的表面张力,N/m;G为Gibbs自由能,J;T为温度,K;p为压力,Pa;As为液体的表面积,m2;n为物质的量,mol。
由此可见,σ是一个与能量相关的物理量,可应用LSERs原理。
已知热力学公式G=U+pV-TS,对于恒温恒压可逆过程,有式(3)存在。

式中,U为体系内能,J;V为体积,m3;S为熵,J/K。
又因为在考虑包括σ在内的广义压力情况下,Gibbs自由能可表示为式(4)。
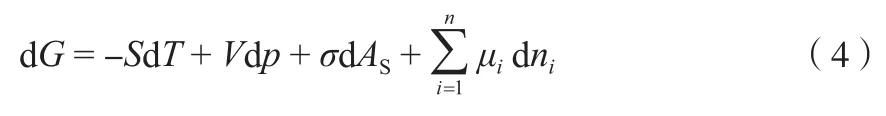
式中,μi为组分i的化学势,J/mol。
运用全微分性质,可得式(5)。

在恒温恒压、体积功为零的情况下,将式(5)代入式(3)可得式(6)。
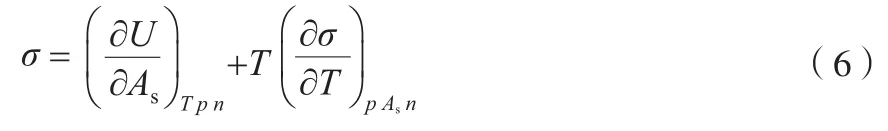
式(6)中右边第一项为增大单位表面积时内能的增量,即为液体内部分子相互作用力的表现。在纯液体中,分子之间的作用力包括氢键作用、偶极-偶极和偶极-诱导-偶极作用、色散力等。因此,根据LSERs理论,将增大单位表面积内能的增量(ΔU)表示为式(7)。

对于式(6)中右边第二项,经实验研究发现,在一定温度范围σ近似为温度的一次函数[19],见式(8)。

因此,由式(6)~式(8)可建立估算纯液体σ的新方程:
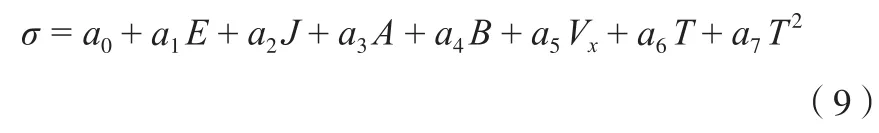
式中,ai为实验数据回归系数。
式(9)中a7T2项是针对某些物质具有接近临界状态的数据而增加的温度校正项。因为当温度趋向临界温度时,σ与温度之间的关系已不遵循线性关系,所以加入该项对温度进行较正,希望提高方程的计算准确度和精确度。
2 结果与讨论
2.1 数据的选取
本工作所选取的分子描述符主要来自Abraham等[16,20-22]发表的文献值。σ数据选自 Jasper等[1-2]整理的实验数据,以及文献[23-24]的实验数据。
2.2 方程相关性分析
选取212种液体有机物,包括烷烃、烯烃、炔烃、卤代烃、芳香烃、醇、醛、酮、酯和有机酸,共2 628个数据点,利用SPSS软件及Excel软件对实验数据及分子描述符进行回归,由最优化结果得到8个关联方程,方程式(9)中各类物质对应系数结果见表1。由表1可知,8个关联方程中各参数与σ的相关系数都达良好级以上,平均相对偏差均低于5%。
对上述各类方程σ数据分别做正态分布,结果见图1。由图1可知,8类有机化合物在临界温度以下范围内的σ估算值的残差都接近正态分布。

表1 计算σ的方程式(9)中各项系数Table 1 Coefficients in the equation (9) for calculating surface tension(σ)
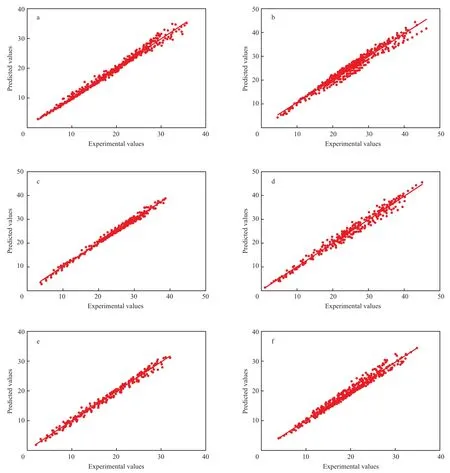

图1 8类有机化合物σ残差的正态分布Fig.1 Normal distribution for σ residuals of 8 kinds of organic compounds.
2.3 方程外推适用性分析
将方程与应用较普遍的Macleod-Sugden关联式和Brock-Bird关联式[3]的计算值相比较,结果见表2。由表2可知,本方程对各类有机物液体σ的预测值与实验值相比,大多数情况下预测偏差都在5%以下,且温度范围较广,有的甚至接近临界温度。本方程估算值偏差在大多数情况下比Macleod-Sugden和Brock-Bird方程的预测偏差小,特别是在高温时。Macleod-Sugden方程和Brock-Bird方程应用于某些种类化合物时会产生较大误差,也就是说这两个方程的普适性受到限制,如Brock-Bird关联式不适用于具有强氢键(极性)的化合物(酸、醇等)。
为了进一步验证方程的外推适用性,另取了8类未参与回归的化合物在298 K时的σ实验数据,与所得方程计算值相比较,结果见表3。由表3可知,用本工作模型对化合物的σ进行估算时,大部分物质的预测偏差都在5%以下。
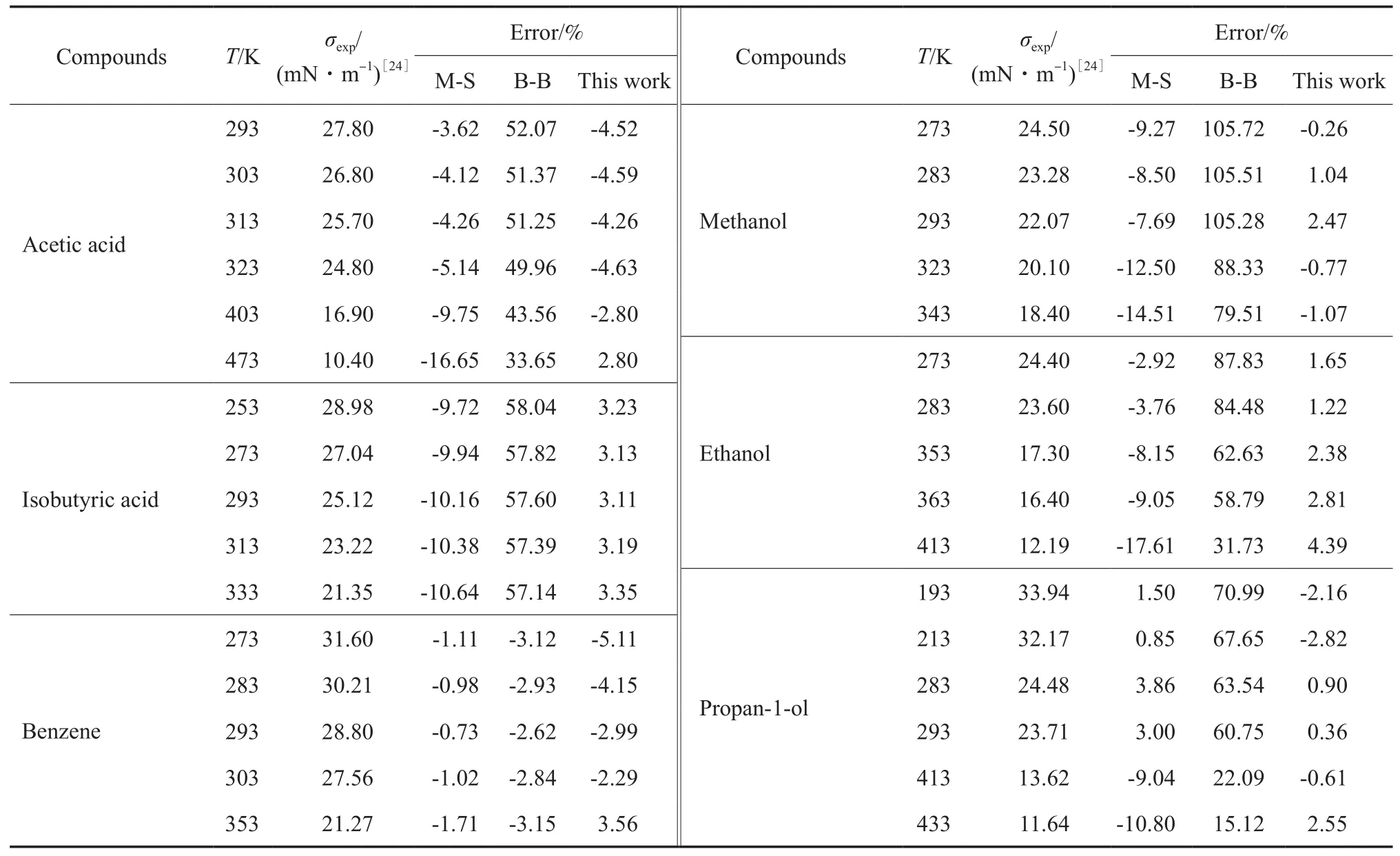
表2 本工作方程与文献方程计算结果比较Table 2 Comparison of calculated results for σ by equations of this work and literature
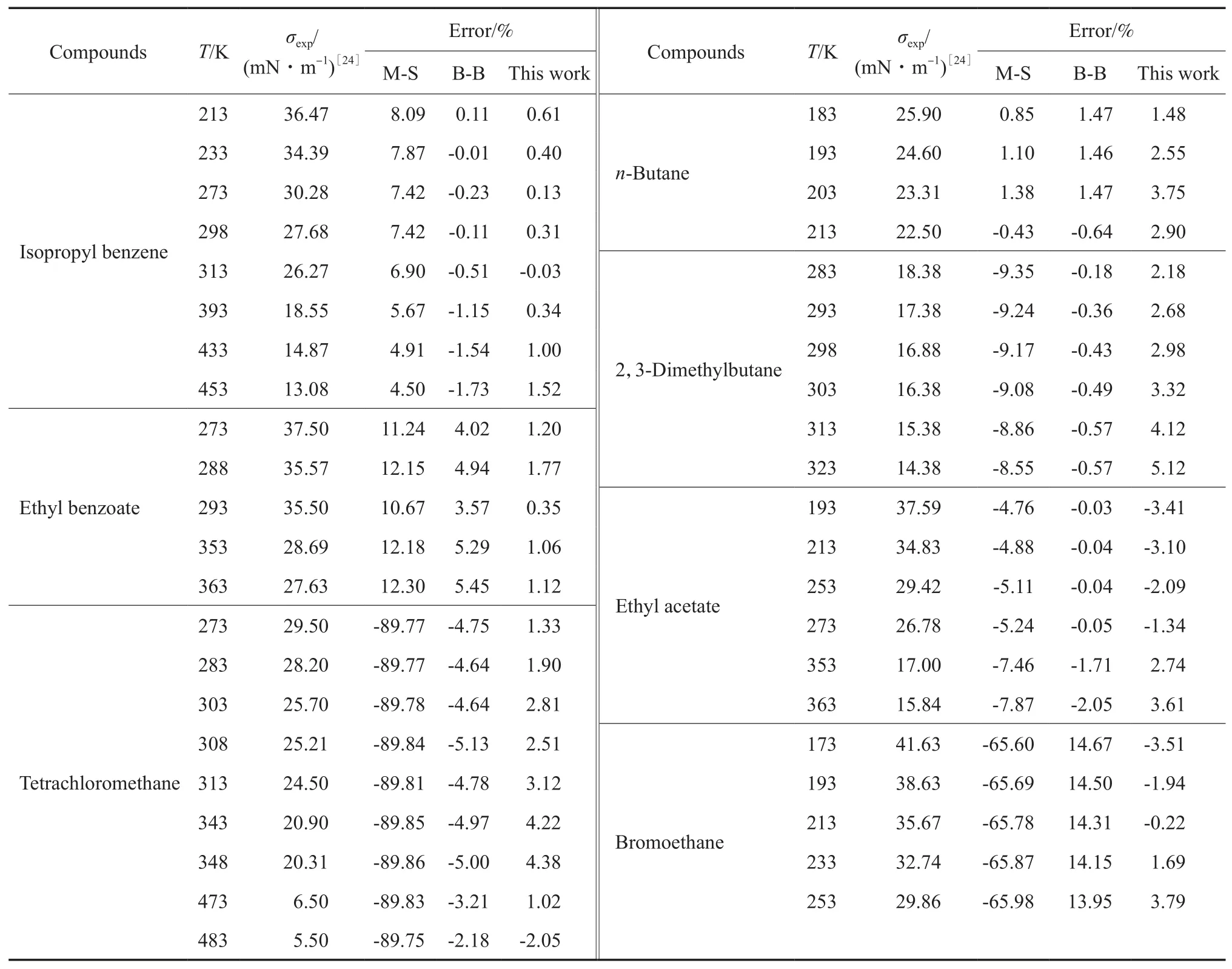
续表2
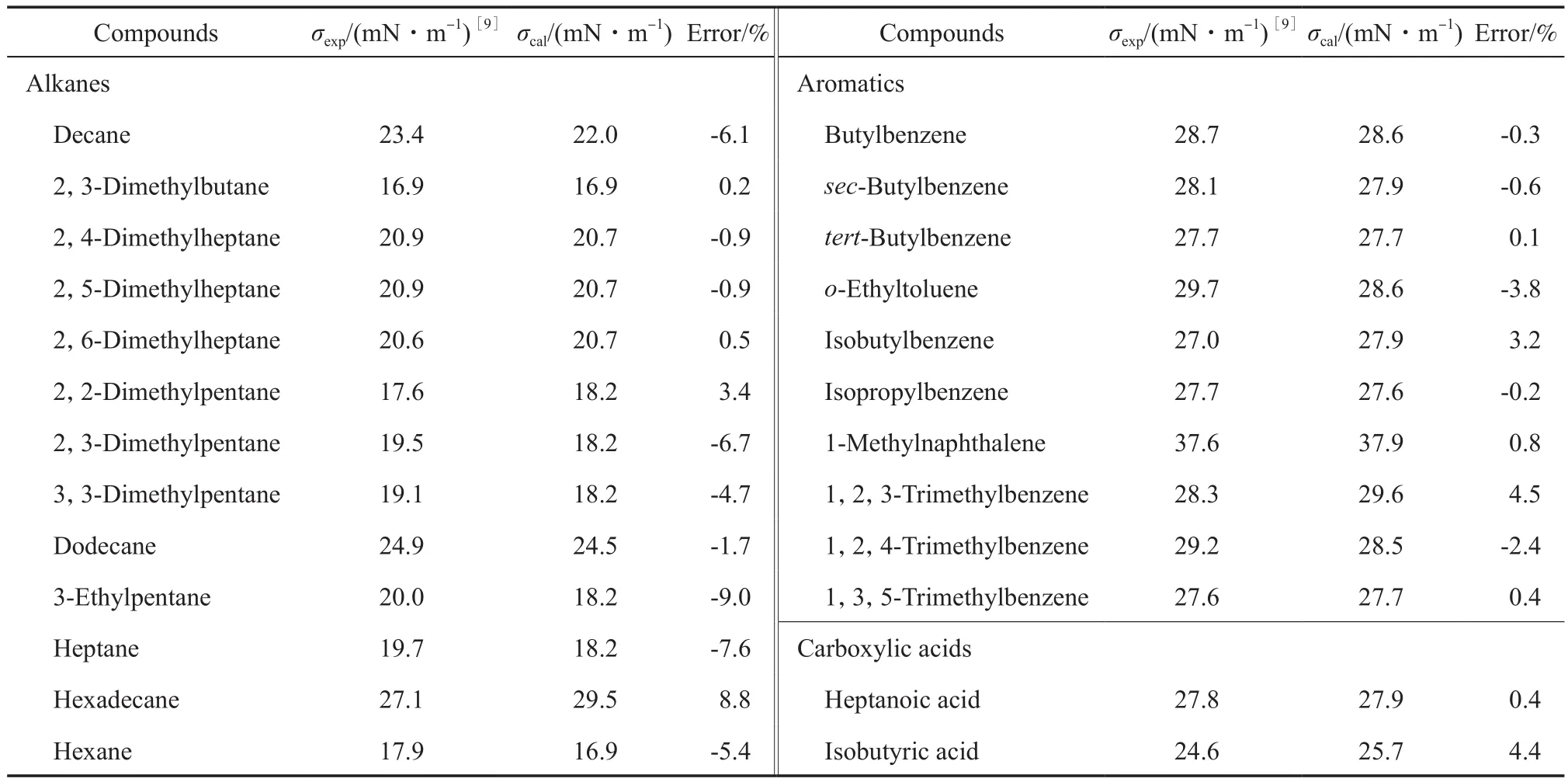
表3 298 K时有机物σ预测结果Table 3 Estimated results for the σ of various compounds by the equation of this work at 298 K
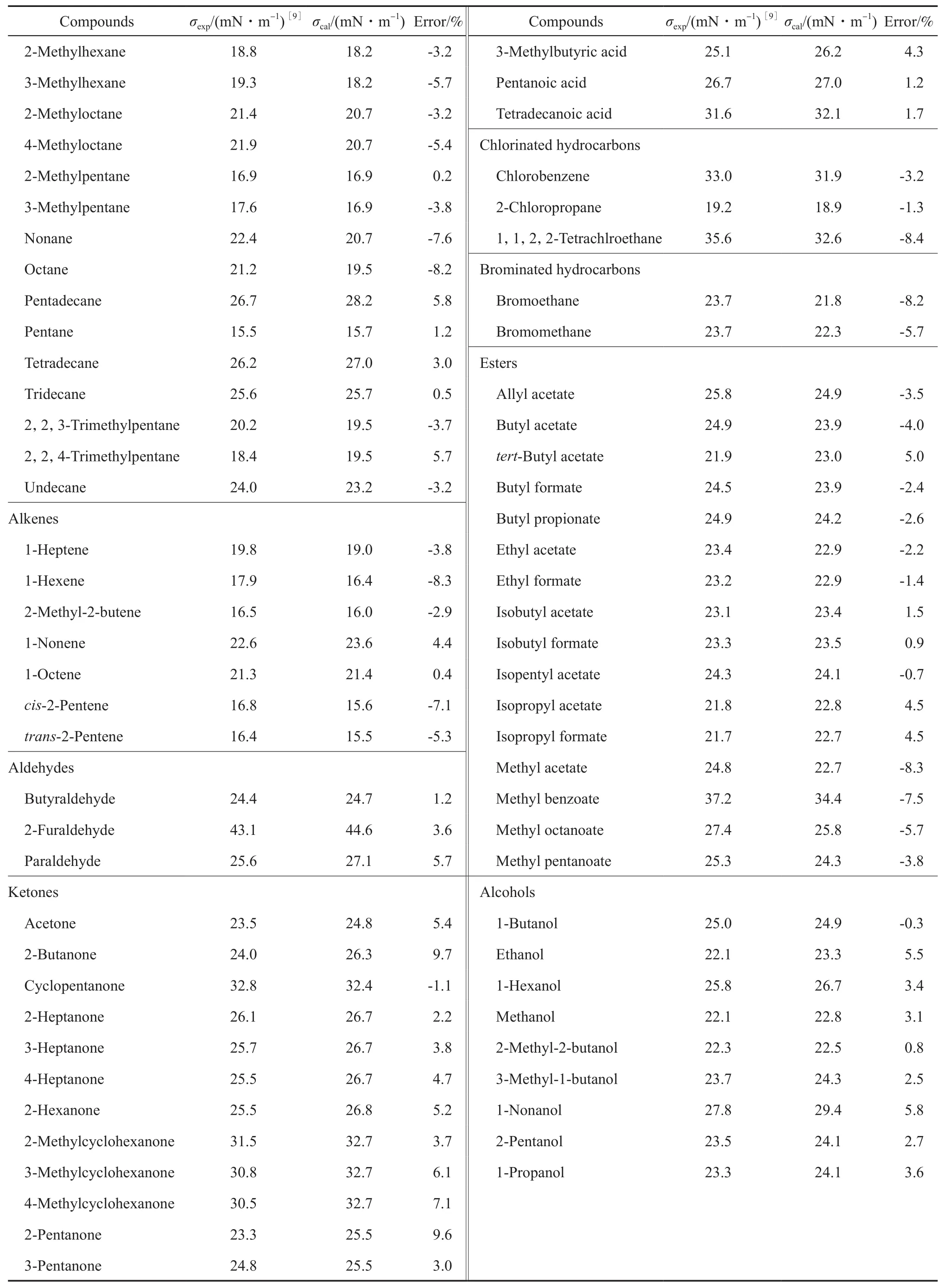
续表3
3 结论
1)提出计算σ的新方法,从本质出发,探讨了分子间相互作用、分子结构对σ的贡献,成功地将宏观物性与微观结构参数相关联,提供了架起宏观与微观桥梁的新方法和经验。
2)提出的计算σ的新方法具有坚实的理论基础,结合应用了基本的热力学公式和LSERs理论,给出的方程形式简单,方程所含参数的物理意义明确、且易于获得,计算方便、快捷,具有高的精度和良好的预测能力,可供工程设计之用。

猜你喜欢
河北师范大学学报(自然科学版)(2022年5期)2022-09-20
作文周刊·小学一年级版(2022年24期)2022-06-18
中学生数理化·八年级物理人教版(2022年4期)2022-04-26
新世纪智能(数学备考)(2021年9期)2021-11-24
波谱学杂志(2021年3期)2021-09-07
中学生数理化·八年级物理人教版(2021年4期)2021-07-22
——以高中化学“氢键”的教学为例
教学月刊(中学版)(2020年13期)2020-12-29
当代陕西(2019年15期)2019-09-02
学苑创造·A版(2018年11期)2018-02-01
小天使·一年级语数英综合(2017年1期)2017-02-16
