
缺陷与掺杂共存的黑磷烯甲醛传感行为的电子理论*
2020-12-14 05:03张国英焦兴强刘业舒张安国孟春雪
物理学报 2020年23期
张国英 焦兴强 刘业舒 张安国 孟春雪
1) (沈阳师范大学物理科学与技术学院, 沈阳 110034)
2) (东北大学生命科学与健康学院, 沈阳 110819)
黑磷烯(black phosphorene, BP)因其“褶皱”的晶格结构而具有较高的比表面积, 在气体吸附及气体传感器方面应用具有很大的优势. 掺杂及缺陷对其传感性有较大的影响. 本文以基于密度泛函理论的第一性原理方法为基础探究了本征、Al 掺杂、含P 原子空缺以及P 空位与Al 掺杂共存的黑磷烯体系吸附甲醛前后的传感行为. 通过建立含缺陷和掺杂吸附体系的结构模型, 计算得出了吸附能、能带结构及电荷转移等电子结构参数. 结果表明, 本征BP 烯以及含P 原子空缺的BP 烯体系对甲醛分子吸附能力较弱, P 原子空缺对电导率以及电荷转移没有影响, 所以本征黑磷烯不适合用于传感器材料. Al 掺杂和P 空位与Al 掺杂共存的BP 烯体系, 吸附甲醛分子的能力明显比前两种情况增强, 电荷转移明显增加, 改变了载流子浓度, 提高了电导率.此外, 在能带图中明显看到产生一个杂质能级, 有效带隙明显变窄, 表明Al 掺杂提高了纯净和含P 空位黑磷烯的传感性. 因此, Al 掺杂和P 空位与Al 掺杂共存的BP 烯体系预计可成为一种新的传感器材料.
1 引 言
随着能源的开发与利用, 一些有毒有害的气体时时刻刻都有可能存在于人们身边. 例如在化学化工、药物制备等工业生产过程中会排放CO2, SO2等有毒有害气体, 家庭装修过程中也会出现甲醛气体超标, 为了安全起见, 利用传感器对有毒有害气体的检测十分必要. 因此开发一种吸附能力强、灵敏度高的气体传感器特别重要. 过去研究者普遍以无机氧化物为材料制备传感器, 由于存在吸附能力弱、灵敏度低等缺陷逐渐被淘汰[1]. 现在研究者大多以纳米材料为核心制作新型气体传感器[2], 例如二硫化物(MoS2和MoSe2)[3]、石墨烯[4−6]和氮化硼(BN)烯[7.8]等. 近年来出现了一种新型二维纳米材料—黑磷烯(black phosphorene, BP), 其结构类似于石墨烯材料的层状结构[9]. 又因为本身“褶皱”的晶格结构而具有更高的比表面积, 在气体吸附以及传感器方面具有很大的优势[10]. 黑磷烯的发现引起海内外学者的广泛研究, 研究结果发现, 纯净的黑磷烯吸附气体的性质不稳定, 因此可以通过掺杂或者结构缺陷的方式提高黑磷烯对气体的反应活性, 达到传感器要求. 例如Kou 等[11]研究发现当钴掺杂的BP 烯吸附一氧化碳(CO)分子时, 吸附能以及电荷转移明显增大, 说明钴的掺杂提高了对CO 分子的传感性. Sun 等[12]应用第一性原理研究了(Li, Al, Ni, Pd, Pt 和Au)金属掺杂对黑磷对CO 分子吸附的影响, 发现金属掺杂可以提高CO 的吸附强度, 金属掺杂的黑磷烯可以作为去除和检测有毒CO 的候选材料. 王晶儒等[13]用密度泛函理论研究了缺陷和S 掺杂对黑磷体系的影响, 结果表明, P 缺陷和S 掺杂都能改变黑磷的能带间隙. 此外, 研究发现黑磷烯在大气中不太稳定, 在氧气和水共存时, 黑磷烯会被腐蚀. 不过黑磷烯可以通过液体剥离法制备表明其具有优异的水稳定性. Xu 和Gao[14]研究了水在黑磷烯表面的吸附, 计算水吸附能为0.086 eV. 而我们前期研究甲醛在黑磷烯表面上的吸附能是0.179 eV[15], 说明甲醛和水分子同时存在时, 甲醛优先吸附于黑磷烯表面. 可见黑磷烯可以用于甲醛的检测. 目前,对本征缺陷与掺杂共存的黑磷烯对有毒或有害气体的吸附行为的研究还较少.
本研究的重点是甲醛(CH2O), 这是在装修过程中产生的对人们身体危害极大的一种有毒气体.因此开发一种吸附能力强、灵敏度高、安全且可靠的气体传感器用于检测甲醛特别重要[16]. 黑磷自发现以来, 大多数研究都集中在其机械和电学性能;关于它的气体吸附特性, 即气体传感性的研究不多. 我们主要是通过使用第一原理方法研究本征缺陷与掺杂共存对黑磷烯吸附甲醛性能的影响. 研究结果表明, 黑磷烯具有作为检测微量甲醛传感器材料的巨大潜力.
2 计算方法与模型
采用基于密度泛函理论的赝势平面波第一原理方法(Material Studio 6.0 软件中的CASTEP软件包[17]), 交换关联能采用广义梯度近似(GGA)[18]对黑磷烯的结构进行优化和性质计算. 在布里渊区积分计算时, 为了确定截断能和k网格设置, 用黑磷体材料进行测试. 同时还比较了考虑自旋极化和不考虑自旋极化的结果, 计算结果列于表1. 从表1可以看出, 不考虑自旋,k网格设置为3 × 3 × 3,增大截断能时, 带隙值增大, 晶格参数a稍有降低,b,c有所增加. 黑磷体材料能隙的实验值约为Eg=0.35 eV[19], 晶格参数的实验值是a= 3.3136,b=10.478,c= 4.3763 Å[20]. 可见k网格设置为3 × 3 × 3,截断能设定为300 eV 比较合理. 截断能取300 eV,k网格设置为2 × 2 × 2, 4 × 4 × 4 时, 带隙值明显与实验值偏离.k网格设置为3 × 3 × 3, 考虑自旋后, 带隙和晶格参数不随截断能发生线性变化.与实验结果相比较发现, 截断能取300 eV 时最好.再比较考虑和不考虑自旋的结果, 发现对于k网格设置为3 × 3 × 3, 截断能设定为300 eV 时, 考虑和不考虑自旋对结果影响不大, 这可能是因为黑磷的P 元素不含d 电子, 本身不存在磁性的缘故. 所以后面的存在空位和掺杂的黑磷烯吸附甲醛的计算考虑到都不存在含d 电子的元素, 所有计算都没有考虑自旋极化的贡献. 优化能量收敛设为10–5eV/atom 以内.

表1 考虑和不考虑自旋极化、取不同截断能和不同k 点时计算的黑磷体材料的能隙和晶格参数Table 1. Calculated energy gap and lattice parameters of bulk black phosphor using different cut-off energies and different k points considering or disregarding spin polarization.
对单层黑磷烯选取1 × 1 × 1, 2 × 2 × 1,3 × 3 × 1 超原胞进行收敛性测试计算. 折合成1 × 1 × 1 超原胞的总能分别为–718.312, –718.578,–718.561 eV. 可见2 × 2 × 1, 3 × 3 × 1 超原胞总能差异在0.01 eV 量级. 为节省计算资源, 单层黑磷烯的超原胞选取2 × 2 × 1, 原胞共20 个P 原子. 为防止BP 层间的相互干扰, 在C方向建15 Å的真空层. 我们优化了纯净的2 × 2 × 1 BP烯的结构,得到其晶格常数为a= 4.41 Å和b= 3.27 Å, 带隙为0.867 eV 与已发表的结果相吻合[21].
缺陷和掺杂的存在会影响材料的稳定性, 其浓度与其在材料中的形成能有关. 通常较高的杂质或缺陷形成能会导致较低的浓度, 同时其稳定性也较差. 黑磷烯BP 及其甲醛吸附体系中P 空位和掺杂Al 的形成能Ef(BP or CH2O/BP: PV),Ef(BP or CH2O/BP: AlP)分别定义为

对于空位和Al 杂质共存的情况, P 空位-Al 杂质对的形成能Ef(BP or CH2O/BP: AlP+PV)由下式给出:
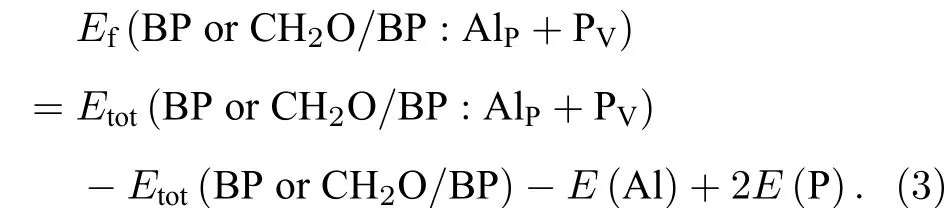
这里,Etot(BP or CH2O/BP: PV),Etot(BP or CH2O/BP: AlP) 是含一个P 空位或一个Al 原子的BP 或CH2O/BP 的总能;Etot(BP or CH2O/BP)是BP 或CH2O/BP 的总能;Etot(BP or CH2O/BP:AlP+PV) 是同时含一个P 空位和一个Al 掺杂原子的BP 或 CH2O/BP 的总能.E(Al),E(P)是金属Al 和黑磷体材料中一个P 原子的能量.
体系的吸附能Ead可以表示为

其中Etot(CH2O) 代表单个甲醛能量;Etot(BP) 代表未掺杂或掺杂后含缺陷BP 体系的能量;Etot(CH2O/BP)表示相应的吸附后体系总能量.
BP 烯吸附体系中, 原子的电荷可以由电荷布居分析求得, 并且体系总电荷由单个原子的电荷叠加得到[22]. 电荷转移ΔQ为
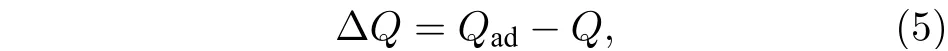
其中ΔQ代表BP 烯吸附体系中电荷转移数;Qad表示为BP 烯吸附的甲醛分子所带电荷量;Q为甲醛气体吸附前所带的电荷量.
气体传感器的工作原理是气体吸附在半导体衬底的表面时, 吸附体系中会发生电荷相互转移,从而引起电导率的变化. 电导率σ 与半导体的带隙Eg之间关系为[23]
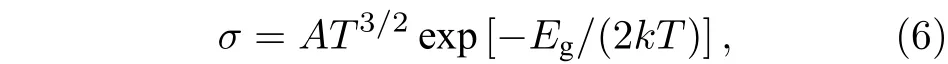
其中A代表常数;T代表温度;k代表玻尔兹曼常数.
3 结果与讨论
3.1 结构性质
缺陷和掺杂的存在会影响黑磷烯的传感性, 也会影响结构的稳定性. 表2 给出了BP 及CH2O/BP体系存在P 空位、Al 掺杂、或P 空位-Al 掺杂对的缺陷形成能. 从表2 可以看出, 黑磷烯中形成Al 替位掺杂比形成一个P 空位要容易得多; 形成替位杂质-空位对比单独形成Al 替位掺杂难, 但比单独形成P 空位容易. 而对于CH2O/BP 体系, 形成掺杂、空位或杂质-空位对都比BP 容易. 当然,由于形成缺陷或掺杂会使黑磷的稳定性有所下降(由于杂质缺陷形成能为正(CH2O/BP 体系Al 替位掺杂除外)).
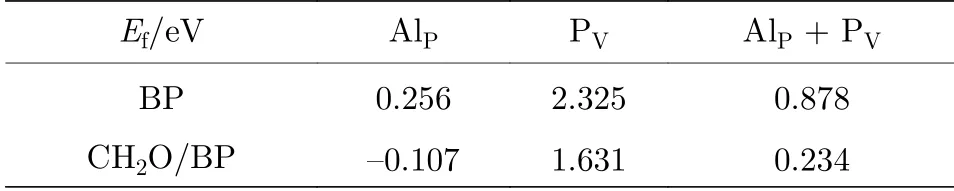
表2 BP 或CH2O/BP 中存在P 空位、Al 掺杂或P 空位-Al 掺杂对时的缺陷形成能Table 2. Defect formation energies of P vacancy, Al impurity, or P-vacancy and Al impurity pair in BP or CH2O/BP.
研究甲醛在本征BP 烯上的吸附时, 要考虑以下几种吸附位. 空位: 在BP 烯体系中空位置; 桥位: 在BP 烯结构P-P 键中间的位置; 顶位: 在BP烯中一个P 原子的顶部正上方的位置. 甲醛分子中的C, H, O 原子分别位于空位、桥位和顶位的正上方. 同时甲醛与BP 烯的平面存在平行、垂直和倾斜三种取向[24]. 对以上每一种情况分别进行几何优化. 结果表明, 当甲醛中的O 原子位于顶位正上方并且倾斜吸附在BP 烯表面时, 此时体系能量最低最稳定, 如图1(a)所示. 优化后结构参数见表3. 甲醛中C=O 键长L(C—O)为1.221 Å, O 原子与BP 烯平面相距D(CH2O—BP) = 3.180 Å. 由(4)式得出体系吸附能为0.179 eV (17.212 kJ/mol< 40 kJ/mol, 说明此时为物理吸附). 根据以上数据可知甲醛气体吸附在本征BP 表面时气敏性很低, 不适合作为传感器材料.

图1 (a) 本征、(b) 掺杂Al、(c) 含P 空位及(d) P 空位与Al 掺杂共存时BP 烯甲醛吸附系统超原胞Fig. 1. The supercells of CH2O/BP adsorption systems :(a) Intrinsic BP; (b) Al doped BP; (c) BP containing P vacancies; (d) BP with the coexistence of P-vacancies and Al doping.
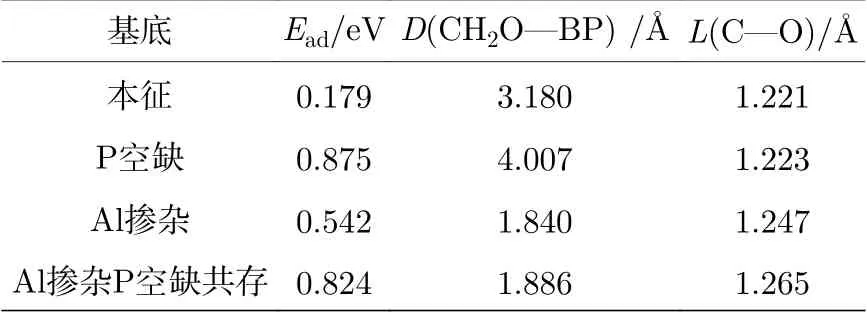
表3 本征、P 空位、Al 掺杂、P 空位与Al 掺杂共存时BP 烯吸附甲醛的优化结构参数Table 3. The optimal structure parameters of intrinsic, P-vacancy contained , Al doping , P-vacancy and Al doping coexisted BP systems adsorbed formaldehyde molecule.
掺杂Al 原子时同样考虑以上三种吸附位置和三种取向. 优化结果表明, 当甲醛分子垂直吸附在BP 烯表面的Al 原子正上方时, 体系能量最低, 达到最稳定状态, 如图1(b)所示. BP 烯存在空位的缺陷结构是指去掉一个P 原子. 因为BP 是褶皱模型, 存在上下两层, 本文选择去掉上层一个P 原子形成空位. 令甲醛分子吸附在空位近邻的P 原子正上方, 因为其他吸附位置经优化发现不稳定. 同时依然让甲醛分子与BP 烯的平面存在平行、垂直和倾斜三种取向. 优化结果表明, 当甲醛中的O 原子位于空位近邻P 原子正上方并且垂直吸附在BP 烯表面时, 体系最稳定, 如图1(c)所示. P 空位与Al 掺杂共存是指去掉一个P 原子后, 在BP 烯上层用一个Al 原子替换一个P 空位近邻的P 原子. 甲醛分子吸附在Al 原子正上方, 同样考虑以上几种吸附位及三种取向, 然后进行结构优化. 优化结果表明, 当甲醛分子的O 原子位于Al 原子正上方(P 原子被Al 取代)并且倾斜吸附在BP 烯表面时, 体系达到最稳定状态, 如图1(d)所示. 根据表3 结构参数可知, 掺杂Al 原子后的BP 烯吸附甲醛分子的吸附能明显大于本征BP 烯的吸附能(Al: 0.542 eV/52.1154 kJ/mol; 本征: 0.179 eV/17.212 kJ/ mol). 当甲醛分子分别吸附在含P 空位的黑磷烯平面上或吸附在含P 空位与Al 掺杂共存的黑磷烯上时, 吸附能都比本征的BP 时的大,(P 空位: 0.875 eV/84.135 kJ/mol; P 空位与Al 掺杂共存: 0.824 eV/79.231 kJ/mol; 本征: 0.179 eV/17.212 kJ/mol). 进一步分析4 种体系吸附甲醛分子时甲醛键长的变化, 结果发现C=O 键长L(C—O)有所增大, 不过含P 原子空缺体系对C=O 键长几乎没什么影响. Al 掺杂体系,D(CH2O—BP)由3.180 Å缩小为1.840 Å, P 空位与Al 掺杂共存的BP 烯体系,D(CH2O—BP)缩小为1.886 Å, 表明Al 掺杂增强了甲醛与BP 烯的相互作用, 验证了吸附能增大观点. 对于P 原子空缺BP 烯体系,D(CH2O—BP)为4.007 Å, 比本征的D(CH2O—BP)还要增大,说明只存在空位缺陷的体系对甲醛分子的吸附很弱.
3.2 BP 烯吸附甲醛的电子性质
通过对本征、含P 原子空缺、Al 掺杂以及P空位与Al 掺杂共存的4 种BP 烯体系的态密度和能带图(图2, 图3, 态密度绘图时高斯展宽设为0.05 eV; 能带计算时k网格取15×15×1, 能带计算路径为Г(0, 0, 0)→X(0.5, 0, 0)→M(0.5, 0.5, 0)→Y(0, 0.5, 0)→Г(0, 0, 0))的计算, 研究了4 种情况下吸附甲醛的电子性质和传感性质. 能量零点表示费米能级的位置, 图中用虚线标出.
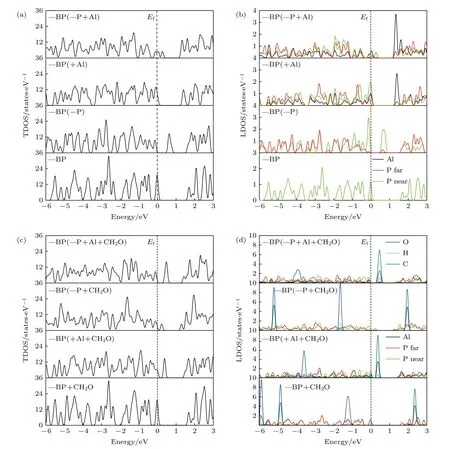
图2 本征、Al 掺杂、含P 空位、P 空位和Al 掺杂共存时吸附或未吸附CH2O 的BP 态密度图 (a), (b) 各BP 烯体系的总态密度和不同原子的局域态密度图; (c), (d) 各吸附CH2O 的BP 烯体系的总态密度和不同原子的局域态密度图Fig. 2. The density of state of intrinsic, Al doped, P vacancy contained, P vacancy and Al doping coexisted BP with or without CH2O adsorption: (a), (b) The total density of state and the local density of state of different atoms of each BP system; (c), (d) the total density of state and the local density of state of different atoms of each BP system with CH2O adsorption.
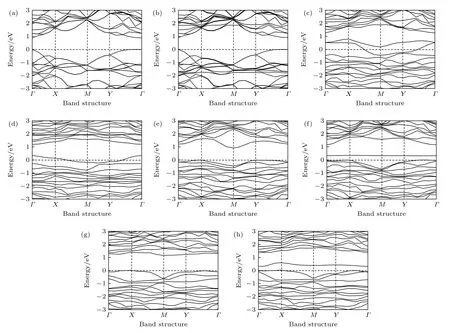
图3 本征、Al 掺杂、P 空位、P 空位和Al 掺杂共存时吸附或未吸附CH2O 的BP 的能带图 (a) 本征BP 烯; (b) 本征BP 烯吸附CH2O; (c) 含P 原 子空位 的BP 烯; (d) 含P 原子 空位的BP 烯吸附CH2O; (e) Al 掺 杂的BP 烯; (f) Al 掺杂BP 烯吸 附CH2O;(g) P 空位和Al 掺杂共存的BP 烯; (h) P 空位和Al 掺杂共存BP 烯吸附CH2OFig. 3. The energy band of intrinsic, Al doped, P vacancy contained, P vacancy and Al doping coexisted BP with or without CH2O adsorption: (a) Intrinsic BP; (b) intrinsic BP after adsorption of CH2O; (c) BP with P atom vacancy; (d) P atom vacancy contained BP after adsorption of CH2O; (e) Al doped BP; (f) Al doped BP after adsorption of CH2O; (g) P vacancy and Al doped coexisted BP; (h) P vacancy and Al doped coexisted BP after adsorption of CH2O.
对于本征BP 烯, 由图2(a)中标(—BP)的部分和图3(a)可知, 此时的费米能级位于价带顶. 图3(a)的能隙为0.867 eV(计算结果), 而态密度图2(a)中显示的带隙明显比能带图中的大. 这是由于在画DOS 图时高斯展宽设为0.05 eV, 如设置为0.1则带隙就很小. 高斯展宽设为0.05 eV 是为了更清楚地显示不同原子对电子结构的贡献, 后面讨论带隙以能带计算为准. 对于本征BP 烯吸附甲醛时,由图2(c)中标(—BP+CH2O)的图像和图3(b)分析可知, 费米能级也处于价带顶, 带隙变为0.891 eV. 由图2(d)中标(—BP+CH2O)的部分图像可以看出, 导带底和价带顶附近的能级是P 原子的贡献, 甲醛分子的各原子在此区域没有贡献. 分析比较上述两种体系下的带隙变化不大(此带隙对应半导体的本征激发), 由(6)式得出纯净黑磷烯体系吸附甲醛前后的电导率变化不大, 因此可知本征BP 烯不适合作为传感材料. 对于含P 空位的BP 烯, 图2(a)中标有(—BP(—P))的是其总态密度图, 可以看出导带底大约在1.5 eV, 价带顶在0 eV 左右. 带隙中存在由P 空位形成的缺陷能级, 在费米能级以上, 且靠近价带, 说明含P 空位的BP 体系是p 型半导体. 图2(b)中标有(—BP(—P))的图是空位近邻和较远P 原子的局域态密度图, 可以看出带隙中央和费米能级处出现了由近邻P 原子贡献的缺陷能级. 由含P 空位的BP 体系的能带图(图3(c))可以发现能带间隙中存在两条缺陷能级并且形成能带, 即最高占据能级和最低未占据能级都由杂质带贡献, 由于最高占据态能带跨越了费米能级, 所以它高于费米能级部分同时成为了最低未占据态能带, 由此可以得出有效能隙为零. 对于含P 空位吸附甲醛的BP 烯, 从图2(c)中标有(—BP(—P+CH2O))的小图得出在费米能级附近能区的电子密度较高, 因此有效带隙几乎为零. 由图2(d)中标(—BP(–P+CH2O))的部分图像可以看出费米能级附近的能级是由P 原子贡献的,甲醛分子的各原子在此区域也没有贡献. 进一步分析能带图(图3(d)), 空位近邻P 原子在费米能级附近能量在–0.2—0.35 eV 之间产生了缺陷能级,由于缺陷能级跨越了费米能级, 所以同样得出有效带隙仍然是零. 由(6)式无法计算得出两种体系的导电率的变化大小, 所以没办法判断是否可以作为传感材料. 对于Al 掺杂的BP 烯, 结合图2(a)中标(—BP(+Al))的小图和能带图(图3(e))分析可知能隙比本征和存在空位的BP 的隙明显变宽,是0.913 eV. 由图2(b)中标(—BP(+Al)的部分图像可以看出价带顶的能级是由Al 和Al 近邻P 原子贡献的, 导带底由Al 原子贡献. 对于Al 掺杂吸附甲醛的BP烯, 由图2(c)和图2(d)中标(—BP(+Al+CH2O))的图和能带图(图3(f))分析得出甲醛在带隙中引入了一个杂质能级, 在价带顶附近, 此时有效带隙为0.315 eV, 对比得出明显小于0.913 eV, 说明掺Al 黑磷烯的带隙明显变小.由(6)式得出引入杂质能级大大提高了的电导率,所以Al 掺杂BP 烯体系对甲醛的传感性大大增加.类似讨论P空位和A l 掺杂共存的BP烯,结合图2(a)中标(—BP(—P+A l))的小图和能带图(图2(g))分析可知能隙是1.153 eV.由图2(b)中标(—BP(—P+A l))的部分图像可以看出价带顶的能级是由A l和P原子贡献的.仔细观察图2(c)和图2(d)中标(—BP(—P+A l+CH2O))的小图和能带图(图3(h))发现P空位和A l掺杂共存的情况与单独掺A l 的BP烯吸附行为类似,只是有效带隙值略有不同,分别约为1.153 eV和0.303 eV,可见P空位和A l 掺杂共存BP烯体系对甲醛的传感性也大大增加.
3.3 BP烯吸附甲醛的电荷分析
甲醛分子及其各原子电荷得失情况见表4.对于本征BP烯体系,当甲醛中的O原子位于顶位并且倾斜吸附在BP烯表面,甲醛的C,O原子与H间产生电荷转移,但是甲醛分子所带总电荷为零,说明甲醛分子与BP烯之间没有电荷转移,说明本征BP烯吸附甲醛前后的导电情况没有变化,并且与甲醛之间也没有离子键相互作用,所以本征BP烯不适合用于传感器制造.含P空位的BP烯体系,甲醛中的O原子位于空位近邻P原子上方并且垂直吸附在BP烯上层表面,甲醛所带的总电荷也为零,说明甲醛分子与含P空位的BP烯之间没有电荷转移.没有电荷转移,说明甲醛吸附前后BP的电导率没有变化,含P空位的BP烯体烯也不能成为传感器材料.当甲醛分子倾斜吸附在掺杂A l和P空位与掺杂A l共存的BP表面的A l 原子正上方时,甲醛分子与掺杂A l 原子BP体系及P空位与掺杂A l 共存BP体系之间电荷转移分别为0.04e和0.15e(甲醛得电子,说明有带正电的空穴注入到黑磷体系中).空穴的注入导致p型载流子密度增大,从而改变了BP烯吸附甲醛时的电导率.这些结果说明A l掺杂或P空位与A l 掺杂共存改善了BP 烯体系对甲醛的吸附性和传感性,这与上面能带结构讨论的结论是一致的.
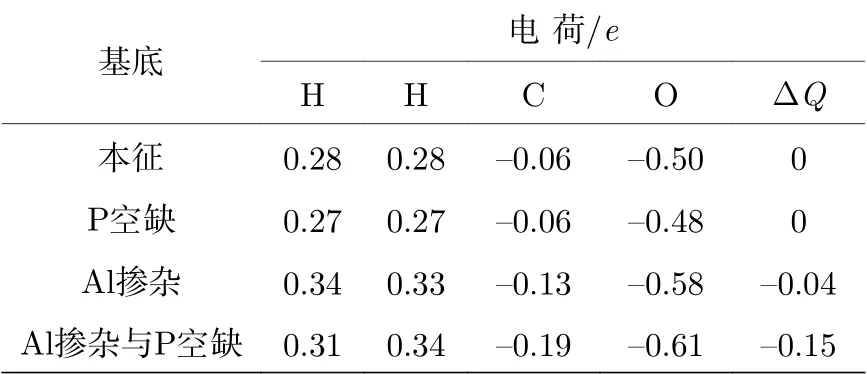
表4 甲醛分子及各原子的电荷得失Table 4.The charge gain or loss of form aldehyde molecu le and its atoms.
4 结论
本文应用第一性原理方法研究了本征、A l 掺杂、含P原子空缺以及P空位与A l 掺杂共存的BP烯体系对甲醛的吸附行为.通过计算得出吸附能、能带结构及电荷转移,分析了不同BP烯体系对甲醛分子的传感机理.研究表明,在本征BP烯上,甲醛分子位于顶位且取倾斜取向;在含P原子空缺的BP烯表面上取顶位垂直吸附;A l 掺杂和在P空位与A l掺杂共存的BP烯上,甲醛位于顶位倾斜取向.本征BP烯以及含P原子空缺的BP烯体系对甲醛分子吸附较弱,电导率以及电荷转移吸附前后没有变化,所以不适合用于传感器材料.A l掺杂和P空位与A l 掺杂共存的BP烯体系吸附甲醛分子的能力明显比前两种情况增强.A l掺杂和P空位与A l 掺杂共存体系吸附甲醛后在禁带中产生了一个杂质能级,使有效带隙比吸附前明显变窄,表明A l 掺杂提高了纯净和含P空位黑磷烯的传感性.因此,A l 掺杂和P空位与A l 掺杂共存的BP烯体系预计可成为一种新的传感器材料.
猜你喜欢
物理学报(2022年17期)2022-09-14
农业工程学报(2022年7期)2022-07-09
电加工与模具(2021年5期)2021-11-05
小天使·聪聪画刊(2021年2期)2021-09-10
汽车零部件(2020年10期)2020-11-09
汉语世界(The World of Chinese)(2019年6期)2019-09-10
作文中学版(2018年1期)2018-11-28
汽车生活(2018年5期)2018-06-21
华东师范大学学报(自然科学版)(2018年2期)2018-05-14
