
黄芩种子萌发期间的代谢变化
2020-12-09 03:30刘金花杨桂玲张永清
园艺与种苗 2020年11期
刘金花,杨桂玲,刘 谦,张永清
(1.济宁市食品药品检验检测中心,山东济宁272000;2.上药控股山东有限公司济宁分公司,山东济宁272000;3.山东中医药大学,山东济南250355)
黄芩为唇形科多年生草本植物黄芩(Scutellaria baicalensisGeorgi)的干燥根及根茎,可清热燥湿、泻火解毒、凉血、止血、除热安胎[1-3],属于大宗常用中药材之一。因功效卓著、临床应用范围广,其市场需求量逐年大幅度增加,仅靠有限的野生资源已经远远不能满足需要,所以近年来有关黄芩的人工栽培研究逐步深入[4-11]。
植物药材的产量与质量最终来源于植物体内的各种生理生化代谢活动。植物体内代谢分为初生代谢和次生代谢。一般来讲,初生代谢形成产量,次生代谢形成质量[12-13]。要达到药材质量稳定可控的目的,需要对植物次生代谢活动的途径、规律及其影响因素进行系统研究。植物次生代谢有多条途径,其中苯丙氨酸途径被认为是最重要的途径之一,因为它在很多天然产物的形成中起着非常重要的作用,比如黄酮类化合物。苯丙氨酸途径的第一步是在苯丙氨酸解氨酶的作用下进行的,它把L-苯丙氨酸转化为反式肉桂酸;第二个重要的酶是肉桂酸-4-羟化酶,它把反式肉桂酸转化成4-香豆酸。查尔酮合成酶是形成黄酮类物质生物合成途径分支上最关键的酶。黄酮类物质是黄芩中的主要活性成分之一,其含量高低与药材质量密不可分[14-16]。研究黄芩植株体内黄酮类物质的生物合成途径及其影响因素,并采取有效措施进行人工调控,对于提高和稳定黄芩药材质量意义重大。种子是新的植物体的雏形体,是植物个体新生命的开始。研究种子萌发期间的代谢变化,特别是活性物质的形成与积累规律,对于探讨植物生长发育过程中药材质量变化具有重要的参考价值。目前,有关黄芩种子代谢方面的研究报道很少,特别是涉及到黄酮类物质形成途径的报道更少。笔者通过种子萌发试验,探讨了黄芩种子萌发期间初生代谢和次生代谢的变化及其相互关系,旨在为探讨黄酮类活性物质的形成规律、控制黄芩药材质量提供理论依据。
1 材料与方法
1.1 试验材料
所用黄芩种子取自山东省日照市莒县库山乡黄芩种植基地,为2 年生植株当年新产种子。经山东中医药大学张永清教授鉴定,确认为唇形科多年生草本植物黄芩(Scutellaria baicalensisGeorgi)的种子。
种子经精选、清洗后,用75%乙醇消毒,再用蒸馏水冲洗干净,放在直径10 cm、铺有3 层滤纸的培养皿中进行发芽。发芽温度控制在25℃,保持湿润。分别在第0、1、2、3、4、5、6、7 天取样,经粉碎提取后,测定各项指标。
1.2 可溶性糖含量测定
采用蒽酮比色法。参照中国科学院上海植物生理研究所[17]的方法,并加以改进。取样品粉末0.1 g,精密称定,分别放入3 支三角瓶中;加入80 mL 蒸馏水,塑料薄膜封口;于超声清洗器中提取30 min,提取液过滤入100 mL 容量瓶中,反复冲洗试管及残渣,定容至刻度;准确吸取样品提取液2 mL 于大试管中,然后按顺序向试管中加入0.5 mL 蒽酮乙酸乙酯试剂和5 mL 浓硫酸,充分振荡,立即将试管置沸水浴中1 min,取出后冷却至室温,以空白作对照,在630 nm 下测定吸光度。根据标准曲线方程计算可溶性糖含量。
1.3 苯丙氨酸解氨酶(PAL)活性测定
参照金丽萍等[18]的方法,称取鲜品0.2 g,研钵预冷,加2 mL 硼酸缓冲液(0.1 mol/L,pH=8.8)和0.02 g 聚乙烯吡咯烷酮,其中缓冲液中含有5 mmol/L 巯基乙醇、1mmol/L EDTA,将样品冰浴研磨成匀浆,在4℃下10000r/min离心15 min,其上清液即为酶粗提液。反应体系中包括2 mL 硼酸缓冲液(0.1 mol/L,pH=8.8)、0.8 mL L-苯丙氨酸(0.02 mol/L)、0.1 mL 酶粗提液,对照不加酶液,改加0.1 mL蒸馏水。30℃下水浴30 min 后,加入0.2 mL 盐酸(6 mol/L)终止反应,于290 nm 波长下测吸光度。按下述公式计算酶的比活力。
酶的比活力[0.01a/(g·h)]=△A×D/0.01×M×T×0.001其中,△A为反应时间内吸光度的变化,M为样品鲜质量,T为反应时间,D为稀释倍数,即提取的总酶液为反应系统内酶液的倍数。
1.4 肉桂酸-4-羟化酶(C4H)活性测定
参照金丽萍等[18]的方法。称取鲜样0.2 g,液氮冷却,加入3 mL 提取溶剂进行研磨。提取溶剂为0.05 mol/L Tris-HCl(pH=8.9),1 mmol/L PMSF,15 mmol/L β-疏基乙醇,10 μmol/L Leupeptin,5 mmol/L VC,0.15% PVP(m/V),4 mmol/L MgCl2,10%甘油。研磨液在4℃下以10 000 r/min离心20 min,取上清液即为C4H 粗酶提取液。酶反应液组成为:0.8 mL 酶提取液,2.2 mL 缓冲液[2 μmol/L 反式肉桂酸,5 μmol/L G6-PNa2,0.05 mol/L Tris-HCl(pH=8.9),2 μmol/L NADP-Na2]。25℃下振荡反应30 min 后,加入100 μL 6 mol/L HCl 终止反应,以10 000 r/min 离心15 min,取上清液在340 nm 处比色测定。参比液为不加酶提取液(加入0.8 mL ddH2O)。
1.5 查尔酮合成酶(CHS)活性测定
参照Zuurbier 等[19]的方法,并加以改进。
1.5.1 查尔酮合成酶粗提液制备。所有操作过程在0℃~4℃环境下进行。将冷冻的10 g 样品放入研钵中,加入少量石英砂和10%(w/w)聚乙烯吡咯烷酮。使用提取缓冲液(0.1 mol/L 磷酸缓冲液,pH 6.8,1.4 mmol/L 2-巯基乙醇,40 mmol/L 抗坏血酸,3 mmol/L 乙二胺四乙酸,10 μmol/L亮抑肽酶和0.2 mmol/L 苯甲基磺酰氯;使用之前先用液氮冷却)将冷冻的粉末混合均匀。待解冻后,在8 000 r/min的转速下离心匀浆20 min,然后将提取的蛋白质使用30%~70%的(NH4)2SO4进行盐析。用2.5 mL 磷酸盐缓冲液(0.1 mol/L,pH 6.8),1.4 mmol/L 2-巯基乙醇,40 mmol/L 抗坏血酸和5%(w/v)海藻糖(使用之前使用液氮冷冻)溶解最后使用70%(NH4)2SO4盐析得到的沉淀,最后用葡聚糖凝胶柱(Sephadex G-25M,Pharmacia)进行脱盐,得到样品。
1.5.2 查尔酮合成酶高效液相定量分析。取500 μL 提取的样品和500 μL 缓冲液[0.5 mol/L 磷酸缓冲液,2.8 mmol/L 2-巯基乙醇,2%(w/v)牛血清白蛋白]并混合均匀。在混合液中加入100 μL 丙二酰辅酶A(0.4 mmol/L)和100 μL 桂皮酰辅酶A(0.2 mmol/L)后反应开始进行,此时需要在30℃的水浴锅中水浴加热40 min。水浴加热之后加入1 mL乙酸乙酯,使用漩涡仪混合均匀,然后离心2 min。将乙酸乙酯层转移到新的容器中,使用真空干燥器挥干乙酸乙酯。再将残余物用300 μL MeOH 溶解,进行高效液相分析,进样体积为20 μL。
1.5.3 高效液相条件。采用Agilent l 100 高效液相色谱仪;Agilent DAD 检测器。洗脱液由MeOH-H2O-85% H3PO4(比例为280∶136∶1,pH 2.6)组成,流速是1.0 mL/min,检测波长为290 nm。
1.6 次生物质含量测定
参照靳维荣等[20]的方法,并加以改进。
1.6.1 仪器条件。Agilent 1100 高效液相色谱仪;Agilent DAD 检测器;Agilent Chemstation 工作站(美国安捷伦科技公司);采用水-甲醇-磷酸溶剂系统梯度洗脱,检测波长为276 nm,流速为1.0 mL/min,柱温30℃,进样体积20 μL。
梯度洗脱时间程序见表1。
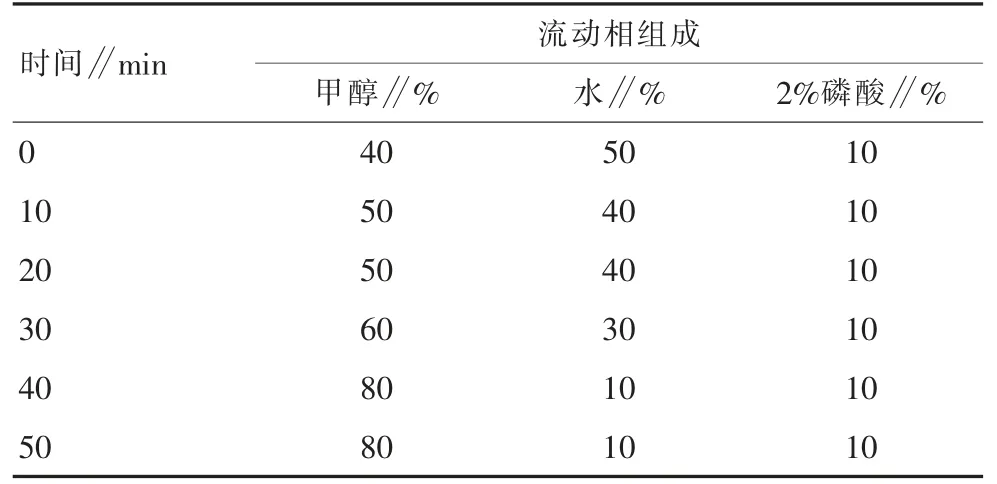
表1 流动相梯度洗脱
1.6.2 对照品溶液制备。分别精密称取黄芩苷、黄芩素、汉黄芩苷、汉黄芩素、野黄芩苷、千层纸素A 对照品6.1、3.0、2.1、1.1、10.3、1.5 mg,用甲醇溶解至100 mL 容量瓶中,混匀。对照品HPLC 图谱如图1 所示。
1.6.3 线性关系考察。取上述黄芩苷、黄芩素、汉黄芩苷、汉黄芩素、野黄芩苷、千层纸素A 混合对照品溶液,分别进样0、2、4、8、12、16、20 μL,按上述色谱条件测定峰面积值。以进样量为横坐标(X),峰面积值为纵坐标(Y),得回归方程分别为Y=90.464X-182.57(R=0.999 1),Y=193.59X-311.68(R=0.999 2),Y=86.437X-37.023(R=0.9992),Y=156.23X-207.14(R=0.9991),Y=49.077X-15.829(R=0.999 3)和Y=1203.84X-110.16(R=0.999 2),表明线性关系良好。
1.6.4 供试品溶液制备。取各黄芩样品0.5 g,精密称定,加入70%乙醇25 mL,称量,超声1 h,放冷,用70%乙醇补足重量,取上清液用0.45 m 滤膜滤过,取续滤液作为供试品溶液。
1.6.5 精密度试验。精密吸取对照品溶液20 μL,连续进样6 次,测定黄芩苷、黄芩素、汉黄芩素、汉黄芩苷、野黄芩苷和千层纸素A 6 种成分的峰面积,其RSD分别为0.23%、0.32%、0.41%、0.29%、0.82%和0.61%,表明精密度良好,符合要求。
1.6.6 稳定性考察。精密吸取试样品溶液20 μL,分别测定0、2、4、6、12、18、24、48、72 h 黄芩苷、黄芩素、汉黄芩苷、汉黄芩素、野黄芩苷、千层纸素A 的峰面积。在72 h 内其RSD值分别为0.86%、0.57%、1.02%、0.93%、1.12%和0.95%,表明供试品溶液在72 h 内是稳定的。
1.6.7 回收率试验。取已知含量的样品0.5 g,精密称定,置25 mL 容量瓶中,分别精密加入黄芩苷、黄芩素、汉黄芩苷、汉黄芩素、野黄芩苷、千层纸素A 对照品溶液,按上述色谱条件进行测定,结果6 种对照品回收率分别是101.22%、99.87%、102.45%、103.21%、98.82%及100.67%,RSD分别是0.63%、0.93%、0.89%、1.29%、1.02%和0.91%。表明6 种对照品回收率符合要求。
2 结果与分析
2.1 萌发期间种子与幼苗的形态变化
图2 结果显示,在试验条件下,第1 天种子吸胀,第3天就开始发芽露出胚根,第7 天形成具备2 片子叶的完整小苗。
2.2 黄芩种子萌发期间可溶性糖的变化
在黄酮类物质的生物合成过程中,可溶性糖是很重要的前体物质。图3 结果显示,随着黄芩种子萌发进程的持续,种子或幼芽中的可溶性糖含量不断降低,在发芽第5 天时达到最低值,但是当幼苗子叶形成时,其可溶性糖含量呈现急剧增加的趋势。在萌发的不同阶段,其可溶性糖含量间均有显著性差异(P<0.05)。这种变化可能与植物的光合作用及淀粉、脂肪类物质的分解有关。当种子萌发时,它自身贮存的营养不断消耗,当子叶展开能进行光合作用后,就会开始光合形成糖类等初生物质。
2.3 黄芩种子萌发期间PAL 和C4H 活性变化
图4 结果显示,萌发伊始黄芩种子中的PAL、C4H 酶活性非常低,近于0,随着萌发进程的持续,种子及幼苗中的PAL 和C4H 活性呈现出逐渐迅速升高的趋势。并且在不同的萌发阶段,其PAL 和C4H 活性均有显著性差异(P<0.05),说明其活性变化非常快。
2.4 黄芩种子萌发期间CHS 活性变化
由图5 可知,CHS 活性变化与PAL、C4H 相似,干种子的活性很低,随着萌发进程的进行,CHS 活性迅速持续升高,不同萌发阶段间差异显著(P<0.05),提示其活性变化也非常迅速。
2.5 黄芩种子萌发期间次生物质含量变化
图6 结果显示,在黄芩种子萌发之前没有检测到次生物质,即没有黄芩苷、黄芩素、汉黄芩苷、汉黄芩素、野黄芩苷、千层纸素A 等黄酮类物质存在。在萌发的第1天,黄芩苷和汉黄芩苷开始积累,但是含量很少,在萌发的第3 天黄芩素、汉黄芩素和千层纸素A 也开始形成了,并且黄芩苷和汉黄芩苷的含量也比第1 天有所增加,在萌发的第5 天野黄芩苷也形成了。所有的次生物质随着萌发的进行都是在不断增加的,特别是在子叶展开时,次生物质含量呈现急剧增加的趋势,但不同的黄酮类物质开始生物合成的时间不同。
2.6 相关性分析
表2 结果显示,在黄芩种子萌发进程中,黄酮类物质含量与PAL、C4H 和CHS 活性呈现出显著正相关,相关系数分别为0.954*、0.987**和0.992**。CHS 与PAL、C4H 也呈现显著性正相关,相关系数分别为0.985*和0.988**。C4H和PAL 也呈现显著性正相关,相关系数为0.988*。由此推测黄酮类物质含量与关键酶活性密切有关,只有酶活性提高后黄酮类物质的含量才能提高。PAL、C4H、CHS 活性间密切相关,说明它们的激活机制具有相同之处。可溶性糖含量的变化是复杂的,其来源是多方面的,所以显示与PAL、C4H、CHS 和黄酮类物质含量间没有显著性相关。
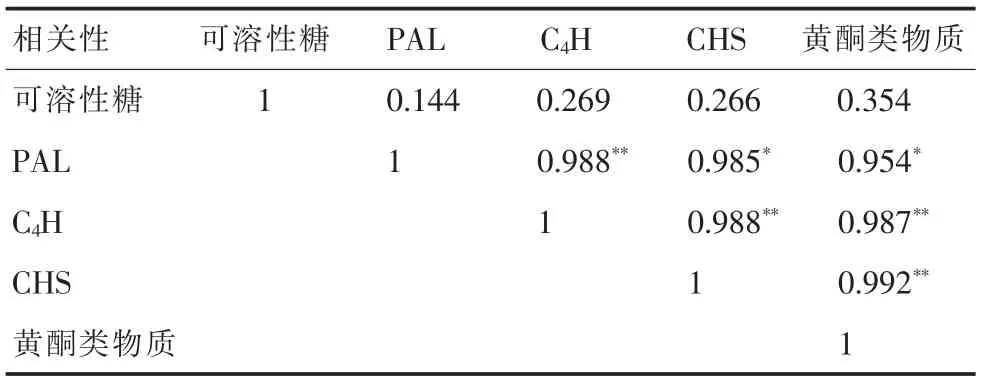
表2 可溶性糖含量、关键酶活性和次生物质含量相关性分析
3 结论与讨论
种子萌发是一个植物新个体生长发育的开始,各种生理生化代谢逐步走向旺盛和复杂,进入了药材产量与质量积累的遗传轨道。黄芩苷、黄芩素、汉黄芩苷、汉黄芩素、野黄芩苷、千层纸素A 等黄酮类物质均具有一定的生物活性,其含量高低在一定程度上代表了药材的质量。该试验结果表明,萌发中的黄芩种子生物代谢激烈,初生物质、次生物质含量出现了显著变化。黄芩种子中不含有任何黄酮类物质,药材质量是在种子萌发后植株生长发育过程中逐步形成和积累的。这一过程既受遗传基因的控制,也受着各种外界因素的影响。各种黄酮类物质的生物合成与积累,首先是在各种酶的催化下进行的,因此酶的合成与活性提高,是各种黄酮类物质合成与积累的前提,酶活性的变化与次生物质含量密切相关。
猜你喜欢
食品工业(2022年5期)2022-06-13
中国药房(2022年10期)2022-05-30
吉林医药学院学报(2022年2期)2022-05-11
中国药学药品知识仓库(2022年5期)2022-04-11
中国药学药品知识仓库(2022年2期)2022-03-23
临床与实验病理学杂志(2021年10期)2021-12-13
中国科技纵横(2021年24期)2021-03-02
今日农业(2020年16期)2020-12-14
蚕桑通报(2020年1期)2020-07-10
分析化学(2018年4期)2018-11-02
