
高效液相色谱法串联质谱法检测南美白虾中呋喃唑酮代谢物的不确定度评定
2020-12-04 02:51蒙丽琼李晨曦梁任佳李学劲冯光毅
现代食品·下 2020年10期
蒙丽琼 李晨曦 梁任佳 李学劲 冯光毅


摘 要:目的:评定高效液相色谱法串联质谱法检测南美白虾中呋喃唑酮代谢物(AOZ)残留量的不确定度。方法:通过分析不确定来源并计算合成不确定度,最终得到南美白虾中呋喃唑酮代谢物的扩展不确定度。结果:当南美白虾中呋喃唑酮代谢物含量为1.93 μg·kg-1时,在95%的置信区间下,扩展不确定度为0.21 μg·kg-1(k=2)。
结论:实验过程的不确定度主要来源于标准曲线拟合过程和标准溶液配制所产生的不确定度。
关键词:高效液相色谱法串联质谱法;南美白虾;呋喃唑酮代谢物;不确定度
Abstract:Objective: To evaluate the uncertainty for the determination of AOZ residue in vannamei shrimp by HPLC-MS. Method: the sources of uncertainty that may be introduced were analyzed and each component of uncertainty was quantified and used to calculate combined uncertainty,and get the expanded uncertainty of AOZ residue in vannamei shrimp. Results: When the total content of AOZ residue in vannamei shrimp was 1.93μg·kg-1, the expanded uncertainty was
0.21 μg·kg-1 (k=2) at a 95% confidence interval. Conclusion: The preparation of standard solution, calibration curve fitting and measurement repeatability were the main sources of uncertainty.
Key words:HPLC-MS; Vannamei shrimp; AOZ; Uncertainty
中图分类号:S948
硝基呋喃类抗生素是具有5-硝基呋喃基本结构的人工合成广谱抗菌药物,硝基呋喃类物质在动物体内迅速分解,原药稳定性只有数小时,呋喃唑酮是硝基呋喃的一种,其在动物体内的代谢物是3-氨基-2恶唑烷基酮(AOZ)。硝基呋喃类代谢物具有细胞诱导性、动物致癌性[1-2]。欧盟1993年将呋喃唑酮禁用于食源性动物,我国也于2002年本部禁用硝基呋喃类抗生素的禁令。在农业部193号公告中,硝基呋喃类抗生素在所有食品动物和所有用途中都被禁止使用[3-4]。然而,显著的临床治疗效果和促生长作用,以及廉价易得的特点,使其仍然存在违法使用现象[5]。因此,动物源性食品中硝基呋喃类代谢物的检测及其不确定的评定具有非常重要的意义。本实验根据国家标准《动物源性食品中硝基呋喃类药物代谢物残留量检测方法》(GB/T 21311-2007)评定南美白虾中呋喃唑酮代谢(AOZ)检测的不确定度。
1 材料与方法
1.1 试剂与耗材
乙腈(HPLC)、甲醇(HPLC)、甲酸(HPLC)、乙酸乙酯(AR)、正己烷(AR)、盐酸(AR)。
呋喃唑酮代谢物标准物质、同位素内标氘代呋喃唑酮代谢物标准物质,来源于农业部环境保护科研监测所。
1.2 仪器和设备
安捷伦6460C列安捷伦液相色谱串联质谱(ESI源);分析天平:SECURA225D-1CN型,检定允许误差为±0.000 05 g;电子天平:JJ1000/0.01 g~1 000 g,
允差±0.05 g。
1.3 方法
1.3.1 样品处理
称取纯2.00 g按照国家标准《动物源性食品中硝基呋喃类药物代谢物残留量检测方法》(GB/T 21311-2007)进行样品处理,过滤膜后,LC-MS/MS检测,内标法定量。
1.3.2 色谱条件
Plus C18色谱柱(4.6×100 mm,1.8 μm);柱温40 ℃;进样量20 μL;流动相A为乙腈,流动相B为10 mmol·L-1乙酸铵水溶液(pH=4.0)。梯度洗脫:
0~3 min时,20%B→40%B,3~6 min时,40%B,
6.01 min时,20%B;流速0.6 mL·min-1。
1.3.3 质谱条件
离子源:ESI(+);扫描模式:MRM;干燥气:温度300 ℃,压力45 psi,流速9 L·min-1;鞘气温度:250 ℃;鞘气流速:11 L·min-1,毛细管电压:4 000 V;定量离子:AOZ为236/134.1,AOZ-D4为240/134;定性离子对:AOZ为236/104。
1.3.4 数学模型
试样中待测组分含量为:
(1)
式(1)中,X-样品中待测物的浓度,单位为
μg·kg-1;C-样液中标准工作曲线所校得到的待测物浓度,单位为μg·L-1;V-样品定容液的体积,单位为mL;m-样品质量,单位为g。
2 结果与分析
2.1 不确定度的来源分析
试样中呋喃唑酮代谢物残留量的不确定度的主要来源有样品质量称量、体积、被测物质量浓度、测量重复性和回收率。
2.2 不确定度的评定
2.2.1 样品称量的不确定度u(m)
称取2 g南美白虾样品,天平最大允许误差为±0.01 g,按照矩形分布s计算,其标准不确定度u(m)和相对标准不确定度urel(m)分别为:
2.2.2 体积产生的不确定度u(V)
样品浓缩后,用2 mL分度吸量管移取2 mL0.1%甲酸水溶解蒸残物。2 mL分度吸量管体积允许误差为±0.012 mL,按照均匀分布计算,其不确定度为:
温度的变化范围为(20±5)℃,水的膨胀系数
β膨胀水为0.001 80 ℃-1,温度变化引起水的体积变化,按照均匀分布计算,产生的不确定度为:
则定容时引入的标准不确定度为:
(2)
2.2.3 被测物的质量浓度产生的不确定度u(C)
C的不确定度由标准系列溶液配制、储备液的稀释以及标准曲线溶液的配制、标准曲线拟合而得。
(1)标准储备液配制引入的不确定度u(C-1)。呋喃唑酮代谢物(AOZ)标准值100 μg·mL-1,扩展不确定度为0.10 μg·mL-1,k=2;呋喃唑酮代谢物-D4溶液标准值50 μg·mL-1,扩展不确定度为3 μg·mL-1,k=2。使用1 mL移液器移取1 mL呋喃唑酮代谢物(AOZ)至10 mL容量瓶中,用乙腈定容配制成
10 μg·mL-1的AOZ储备液,使用1 mL移液器移取1 mL呋喃唑酮代谢物-D4溶液标准物质至50 mL容量瓶中,用乙腈定容配制成1 μg·mL-1的AOZ-D4储备液。
呋喃唑酮代谢物(AOZ)标准品和呋喃唑酮代谢物-D4溶液标准物质引入的相对不确定度为:
(2)标准溶液稀释产生的不确定度u(C-2)。用
1 mL单标线吸量管移取储备液1 mL于10 mL容量瓶(A级)中,用乙腈定容至刻度,摇匀,得到10 μg·mL-1的标准溶液(1∶10稀释)。按照均匀分布处理,玻璃器具及温度波动引入的不确定度见表1。
则由AOZ储备液由100 μg·mL-1稀释到
10 μg·mL-1过程产生的相对标准不确定度为:
同理,再逐级稀释到100 ng·mL-1,则得到AOZ储备液稀释过程产生的相对标准不确定度为:
则由AOZ-D4储备液由50 μg·mL-1稀释到
1 μg·mL-1过程产生的相对标准不确定度为:
同理,再逐级稀释到100 ng·mL-1,则得到AOZ-D4储备液稀释过程产生的相对标准不确定度为:
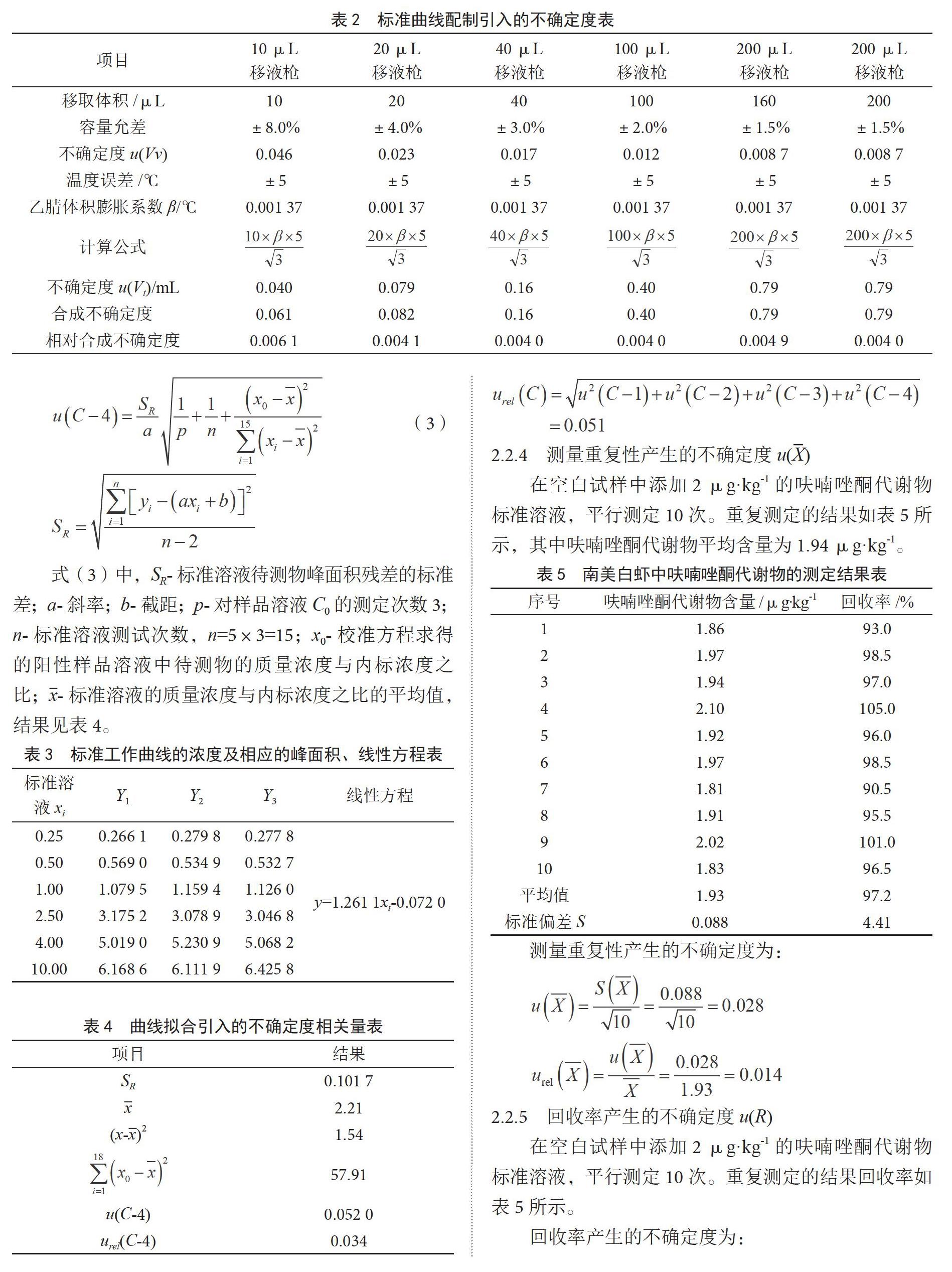
(3)标准曲线配制过程产生的不确定度u(C-3)。
使用20、100、200 μL移液器分别移取适量的
100 ng·mL-1 A0Z标准溶液加入空白南美白虾基质中,按照样品处理,最终用2 mL单标移液管加入2 mL 0.1%甲酸水定溶液,得到质量浓度分别为标准系列工作溶液。移液器的刻度误差参考《移液器检定规程》
(JJG 646-2006)的要求,按A类评定,如表2所示。2 mL单标移液管的不确定度见(2)urel(V)=0.006 0。
则标准系列溶液配制过程引入的相对标准不确定度为:
(4)标准工作曲线拟合产生的不确定度u(C-4)。将标准系列溶液,分别重复进样3次,以峰面积(Ai)与内标峰面积(As)之比为纵坐标yi,标准溶液浓度(Ci)与内标浓度(Cs)之比为横坐标xi,采用最小二乘法拟合而成的线性回归方程为yi=axi+b,测定数据及计算结果见表3。取一阳性样品,对各待测化合物浓度C0进行3次重复测定,其待测物浓度与内标浓度之比的平均值为0.971,则由标准曲线拟合产生的不确定度按(3)式计算:
(3)
式(3)中,SR-标准溶液待测物峰面积残差的标准
差;a-斜率;b-截距;p-對样品溶液C0的测定次数3;
n-标准溶液测试次数,n=5×3=15;x0-校准方程求得的阳性样品溶液中待测物的质量浓度与内标浓度之比;x-标准溶液的质量浓度与内标浓度之比的平均值,结果见表4。
2.2.4 测量重复性产生的不确定度u(X)
在空白试样中添加2 μg·kg-1的呋喃唑酮代谢物标准溶液,平行测定10次。重复测定的结果如表5所示,其中呋喃唑酮代谢物平均含量为1.94 μg·kg-1。
测量重复性产生的不确定度为:
2.2.5 回收率产生的不确定度u(R)
在空白试样中添加2 μg·kg-1的呋喃唑酮代谢物标准溶液,平行测定10次。重复测定的结果回收率如表5所示。
回收率产生的不确定度为:
2.3 合成不确定度
由公式计算可得呋喃唑酮代谢物的合成不确定
度为:
2.4 扩展不确定度及结果表示
依据JJF 1135—2005,呋喃唑酮代谢物含量的扩展不确定度U(X)=urel(X)×X×2为0.21 μg·kg-1,故液相色谱串联质谱法测定南美白虾中的呋喃唑酮代谢物含量的结果(1.93±0.21)μg·kg-1,k=2。
3 结论
经实验发现,标准曲线拟合过程和标准溶液配制所产生的不确定度最大,其次为回收率、测量重复性的影响。在检测过程中,可通过严谨配制标准曲线,增加平行样品测定次数,提高检测人员的操作数练程度,可以降低不确定度,提高检测结果的准确性。
参考文献:
[1]岳振峰.食品中兽药残留检测指南[M].北京:中国标准出版社,2010.
[2]徐伟,耿士伟,刘路,等.硝基呋喃类药物及其代谢物检测方法的研究进展[J].天津农业科学,2018,24(8):16-20.
[3]农业部.动物性食品中兽药最高残留限量[Z].2002-12-24.
[4]中华人民共和国农业农村部,国家卫生健康委员会,国家市场监督管理总局.GB 31650-2019 食品安全国家标准 食品中兽药最大残留限量[S].北京:中国标准出版社,2019.
[5]中华人民共和国农业农村部.关于《2019年国家产地水产品兽药残留监控计划》上半年实施情况的通报[Z].2019-09-09.
猜你喜欢
三农资讯半月报(2020年19期)2020-10-27
科技创新导报(2020年5期)2020-06-11
中国食品(2020年9期)2020-05-26
中小企业管理与科技·下旬刊(2019年3期)2019-07-08
食品安全导刊(2017年12期)2018-01-04
科教导刊(2017年26期)2017-11-07
食品界(2017年7期)2017-08-24
科技与创新(2015年17期)2015-09-11
热带农业科学(2015年4期)2015-06-18
分忧(2015年3期)2015-06-08
