
玉米雄性不育突变体mi-ms-3的遗传分析及分子鉴定
2020-11-27 13:23:40田士可秦心儿张文亮代明球
作物学报 2020年12期
田士可 秦心儿 张文亮 董 雪 代明球 岳 兵
研究简报
玉米雄性不育突变体的遗传分析及分子鉴定
田士可 秦心儿 张文亮 董 雪 代明球 岳 兵*
华中农业大学作物遗传改良国家重点实验室, 湖北武汉 430070
玉米是杂种优势利用的最好的作物之一, 雄性不育材料作为一种宝贵的种质资源, 对杂种优势的利用具有十分重要的价值。前期筛选得到1个雄性不育突变体, 并将其命名为。该突变体表现为: 雄穗花药数目减少且不外露, 每个花药只有2个药室, 部分花药退化为膜状并在其末端形成丝状物; 1% I2-KI染色发现花药中含有能正常着色的花粉粒, 与野生型花粉镜检不同之处在于总花粉粒数目的减少; 突变体雌穗花丝增多, 成熟果穗的籽粒两侧各有1个败育的籽粒。通过与自交系Mo17杂交得到F1完全正常。利用F2分离群体进行遗传分析发现突变表型由隐性单基因控制。利用BSA法, 初步将该基因定位在3号染色体长臂上。随后利用29对SSR标记和10对Indel标记, 将突变基因定位到S-6和umc1027两个标记之间, 物理距离为1.5 cM。该定位区间内有21个候选基因, 通过转座子标签法及测序分析, 最终发现()基因ATG上游30 bp处发生了转座子的插入突变。分析表明,与之前所报道的由于碱基突变造成的突变体的突变方式不同, 是一个新的的等位突变体。RT-PCR分析表明基因在突变体中表达量降低。的发现为玉米花器官发育研究以及不育化杂交制种提供了新材料。
玉米; 雄性不育; 基因定位; 遗传分析
植物雄性不育是指雄性器官不能正常发育, 产生功能异常的雄配子, 但雌性器官能够正常发育, 可以与其他有功能的雄配子受精结实, 且植株不育性能够稳定遗传的现象。很多原因都会导致雄性不育现象的产生, 如基因漂移、基因突变、光照周期及温度的改变等[1]。雄性不育按照遗传方式的不同, 可以分为3类: 细胞质雄性不育、细胞核雄性不育以及核质互作雄性不育, 其中细胞核雄性不育还分为显性核不育和隐形核不育, 且以后者为主。
花器官作为高等植物的主要特征, 其发育模型由经典ABC模型逐步完善为之后的ABCD模型、ABCDE模型及四因子模型。玉米具有2类单性花序, 雄穗和雌穗作为成熟花的基本单位分别位于顶生花序和侧生花序上[2]。目前在玉米中已经鉴定出多个花器官发育突变体, 这些突变体对玉米花器官发育机理的研究、遗传育种的应用和提高杂种优势利用效率具有重要价值。玉米中B类基因有()、、/()、和[3]。是拟南芥同源基因, 它在花药和浆片原基中表达, 在突变体中心皮替代雄蕊, 稃状结构替代浆片[4]。、、和是拟南芥同源基因。()突变体的雄穗及浆片转化为内外稃结构, 同时本应在雌穗发育过程中退化的雄蕊转化为心皮结构, 导致雌穗花丝增多。
通过创制玉米雄性不育系对大程度的降低人工成本、提高作物育种效率及种子纯度具有重要意义。本研究在B73与::突变体杂交后代中筛选得到1个雄性不育突变体, 针对该突变体进行遗传分析及基因定位, 并进一步对该突变体在杂种优势中的利用进行了探讨。
1 材料与方法
1.1 试验材料
试验材料包括玉米自交系B73、Mo17和::突变体(美国玉米种质中心引进)。以B73与::突变体杂交, 杂交后代经多代自交筛选得到雄性不育突变体, 将该不育材料命名为雄性不育系。上述材料均种植于华中农业大学校内试验基地。
1.2 突变体的表型鉴定和遗传分析
野生型和突变体的表型调查情况如下: 散粉情况在雄穗开花后第2天上午9:00—12:00进行统计, 并根据散粉能力分为4个等级(0级, 不散粉; 1级, 少量散粉; 2级, 较多散粉; 3级, 正常散粉); 花药外露在雄穗开花结束后进行调查(突变体植株, 在其雌穗吐丝2~3 d后进行统计), 根据花药外露情况分为4个等级(0级, 不外露; 1级, 外露少于1/2; 2级, 外露多余1/2; 3级, 全部外露); 部分植株花药退化形成丝状突出物, 对雄穗上有丝状突出物的植株进行统计; 采用1% I2-KI对花粉染色进行育性调查, 每株重复3次; 取雄穗中部小穗, 剥离上位花花药, 用卡诺氏固定液固定, 送拜意尔生物公司进行花药切片观察。
以为母本, Mo17为父本, 杂交得到F1代, F1自交获得F2分离群体。对F2群体进行遗传分析, 在散粉期调查各单株表型, 统计各群体总株数、可育株数及不育株数, 利用卡方测验分析遗传模式。
1.3 目的基因初定位
构建的F2群体中于散粉期分别选取30株可育株和不育株, 提取DNA用于构建“野生型池”和“突变体池”, 利用BSA (Bulk Segregant Analysis)方法对进行初定位。根据定位结果在MaizeGDB数据库(http://www.maizegdb.org/)中选取多对的SSR标记(表1), 另外12份可育株和不育株组成的“野生型池”和“突变体池”进行聚丙烯酰胺凝胶电泳, 进行SSR标记多态性筛选及突变位点定位。
1.4 DNA的提取、分子标记的开发及候选基因预测
在抽雄前完成对亲本和分离群体取样, CTAB法提取植株DNA[6]。待散粉结束完成育性调查后, 用于基因定位。根据初步定位结果, 在MaizeGDB上下载B73参考基因组序列, 用Blast与Mo17序列进行比对, 找出单拷贝区段中有InDel差异的位点, 根据这些差异位点设计显性标记(表1), 来源于Mo17的序列可以扩增出产物, 而来源于B73的序列则不能, 再通过琼脂糖凝胶电泳来筛选交换单株, 并通过标记加密进一步缩小定位区间。
由于该突变表型是转座子发生转座造成插入突变导致, 且大多数转座子末端包含反向重复序列, 因此利用转座子末端反向重复序列来设计引物[7], 通过该引物与候选基因特异性引物组合扩增的方法来确定候选基因。
1.5 PCR扩增及序列分析
利用Primer 3在线设计引物, 由武汉天一辉远生物科技有限公司合成引物。PCR反应总体积为15 μL, 含2 × PCR Mix 7.5 μL、模板1.5 μL (50 ng μL–1)、引物各1 μL (5 μmol L–1), ddH2O补至15 μL。PCR反应条件为预变性(94℃, 5 min); 变性(94℃, 30 s), 退火(根据m值而定, 30 s), 延伸(72℃, 1 min), 34个循环; 72℃延伸5 min。
在EnsemblPlants网站(http://plants.ensembl.org/ index.html)上下载目的基因ATG上游2500 bp序列, 用启动子元件预测网站PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/)预测目的基因的启动子区调控元件, 对PlantCARE预测结果进行分析整理, 再利用TBtools软件[8]对结果进行可视化分析。
1.6 RNA提取及表达量分析
取抽雄前雄穗幼穗, 一部分放入液氮中速冻后于-80℃保存, 另外一部分用于表型观察, 待后期表型和基因型鉴定后, 挑选出相应的野生型和突变体提取RNA。参照TRI pure Reagent总RNA提取试剂盒(北京艾德莱生物科技有限公司)操作手册提取RNA, 参照反转录试剂盒(北京全式金生物技术有限公司)说明书合成cDNA。采用RT-PCR检测目的基因在野生型和突变体中的表达量。
2 结果与分析
2.1 mi-ms-3的表型鉴定
突变体植株在营养生长阶段与野生型植株无差异; 而在生殖生长阶段, 突变体植株虽能正常抽雄, 但花药基本不外露(图1-A)。肉眼及花药石蜡切片观察发现, 突变体花药瘦小, 只有2个药室, 其余2个药室退化(图1-B1, B2)。突变体雄穗花药数目与野生型相比, 下位花无花药,上位花有0~3数目不等的花药, 部分花药退化为膜状, 并在其末端形成丝状物(图1-C1~C4)。1% I2-KI染色发现, 突变体花药中包含花粉, 且花粉粒能正常着色(图1-D1, D2), 但花药不开裂、不散粉。与野生型植株雌穗相比, 突变体雌穗在开花期表现为花丝增多, 每个子房对应2~4个花丝(图1-E1, E2); 授粉后, 突变体成熟果穗的籽粒两侧存在败育的籽粒。

表1 基因定位及基因表达分析的引物

图1 mi-ms-3和野生型表型
A: 野生型(左)和(右)雄穗表型,花药不外露; B1~B2: 花药发育后期, 野生型和花药横切片,花药只有2个药室; C1~C4:突变体小穗, 分别包含0~3个花药; D1~D2: 野生型和花粉粒; E1:(左)和野生型(右)雌穗表型,雌穗花丝增多; E2:(左)和野生型(右)籽粒表型,每个籽粒对应多个花丝。
A: the tassel of WT(wild type) (left) and(right),anther not exserted; B1–B2: transverse sections of the entire anther of WT andin the late microspore developmental stage,anthers with only two pollen sacs; C1–C4: the spikelet ofcontain 0–3 anthers respectively; D1–D2: pollen grains of WT and; E1: the ear of(left) and WT (right), the number of silks in the ear of theincreased; E2: the kernel of(left) and WT (right),each seed corresponds to multiple silks.
2.2 mi-ms-3的遗传分析
通过对突变体与Mo17杂交得到的F2分离群体进行遗传分析发现, F1代雄穗与雌穗均表现正常, F2群体野生型和突变体单株分别为73株和30株, 符合3﹕1 (c2= 0.94,= 0.33), 说明该突变表型是由隐性单基因控制。
2.3 突变基因的定位和克隆
利用本实验室已合成的全基因组SSR引物对构建的2个极端混池进行扩增, 筛选多态性标记, 并初步将目的基因定位在3号染色体长臂上。根据育性调查结果, 在F2群体中随机选取野生型和突变体各12株, 构成另一组混池。在MaizeGDB数据库中选取29对SSR标记在“野生型池”和“突变体池”之间进行多态性标记筛选。对聚丙烯酰胺凝胶电泳结果分析发现, 其中8对SSR标记在“野生型池”和“突变体池”中具有多态性, 用这8对SSR标记对分离群体进行检测, 初步将目的基因定位在 umc1973和umc1027两个SSR标记之间, 物理距离为5.9 Mb。
为了进一步精细定位, 根据B73和Mo17参考基因组在umc1973和umc1027两个标记间的序列差异开发InDel标记, 并对2个亲本进行多态性分析, 共筛选出10对Indel标记(表1), 最终将候选基因定位到S-6和umc1027两个标记之间, 物理距离为1.5 cM (表2)。对定位区间内的21个基因进行分析, 根据基因表达模式, 选择在花药中表达量较高的基因作为候选基因, 包括、、、和。根据转座子标签法, 通过利用转座子末端反向重复序列设计的引物TIR-1与候选基因特异性引物42618-R组合进行PCR扩增,最终确定发生插入突变的基因为(图2), 该基因编码MADS-box蛋白, 是参与花器官发育模型的B类功能基因。通过对野生型和突变体中该基因全长扩增比对发现, 突变体中该基因扩增片段长度要比野生型长。测序分析表明, 在基因起始密码子ATG上游30 bp处有1494 bp序列插入, 在插入位点处形成9 bp靶位点重复(TTCCTTGGG) (图3)。对1494 bp序列进行分析发现, 其为1个完整的转座子, 该转座子具有转座活性。
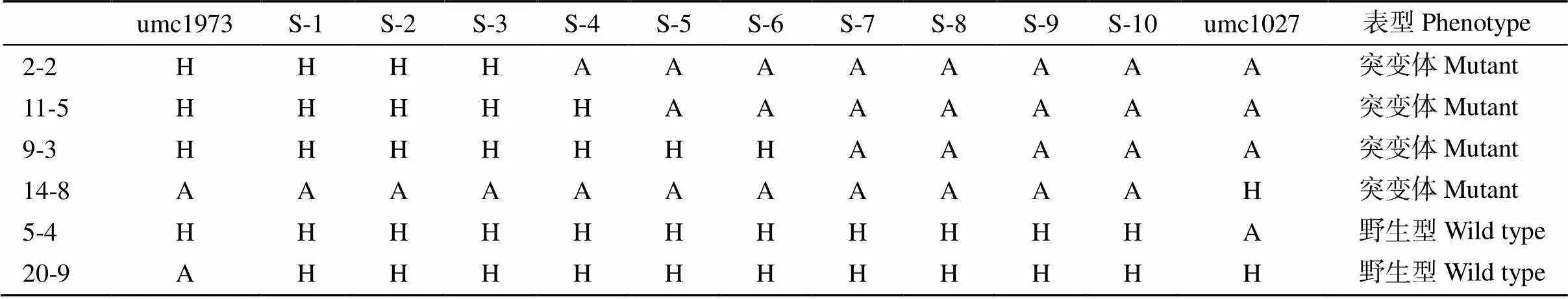
表2 重要重组单株基因型及表型
H表示杂合基因型; A表示突变体基因型。
H denotes the heterozygosity genotype; A denotes the genotype of mutant.
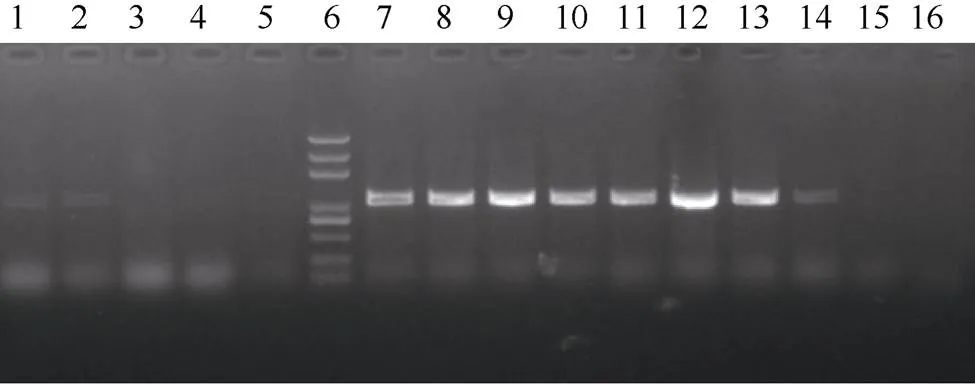
图2 Mu转座子插入突变体的鉴定
1~2: 杂合野生型植株; 3~5: 纯合野生型植株; 6: marker; 7~14: 纯合突变体植株; 15: B73; 16: Mo17。
1–2: heterozygous plants; 3–5: homozygous wild type plants; 6: marker; 7–14: homozygous mutant plants; 15: B73; 16: Mo17.
2.4 zmm16的启动子分析
启动子预测分析结果显示,基因的启动子区包括茉莉酸甲酯响应元件、参与防御和应激反应响应元件、生长素响应元件、脱落酸响应元件、干旱胁迫响应元件及厌氧响应元件(图4)。研究表明, 茉莉酸甲酯响应元件与玉米雄穗和雌穗的发育有关[9], 特别是与玉米单性花形成密切相关[10]。除此之外,基因启动子区还存在与分生组织表达相关的顺式作用元件, 这也进一步说明转座子插入是导致花药发育异常的原因。另外, 还发现低温响应顺式作用元件及多个光响应元件, 这2类响应元件对玉米基因功能的影响值得在未来深入探讨。
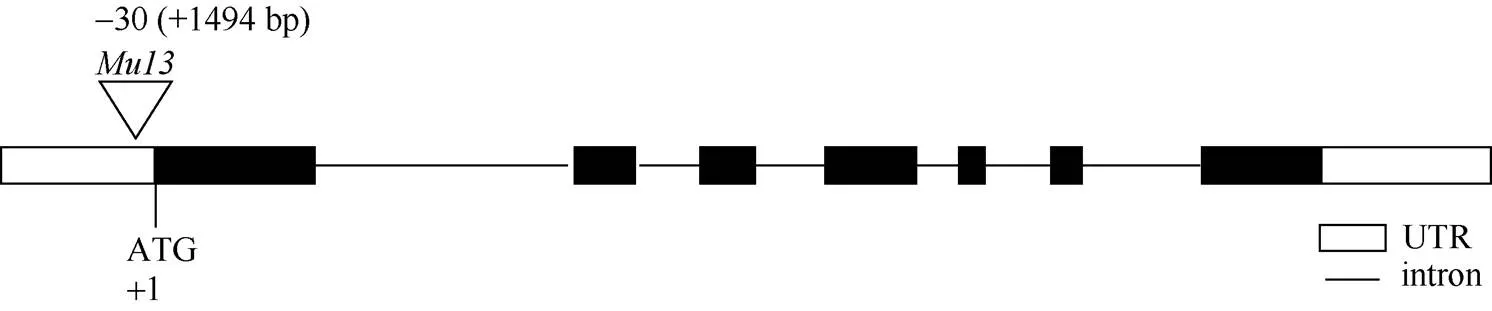
图3 zmm16基因序列分析
转座子插入到基因ATG上游30 bp处。transposon occurred at 30 bp upstream of ATG of.
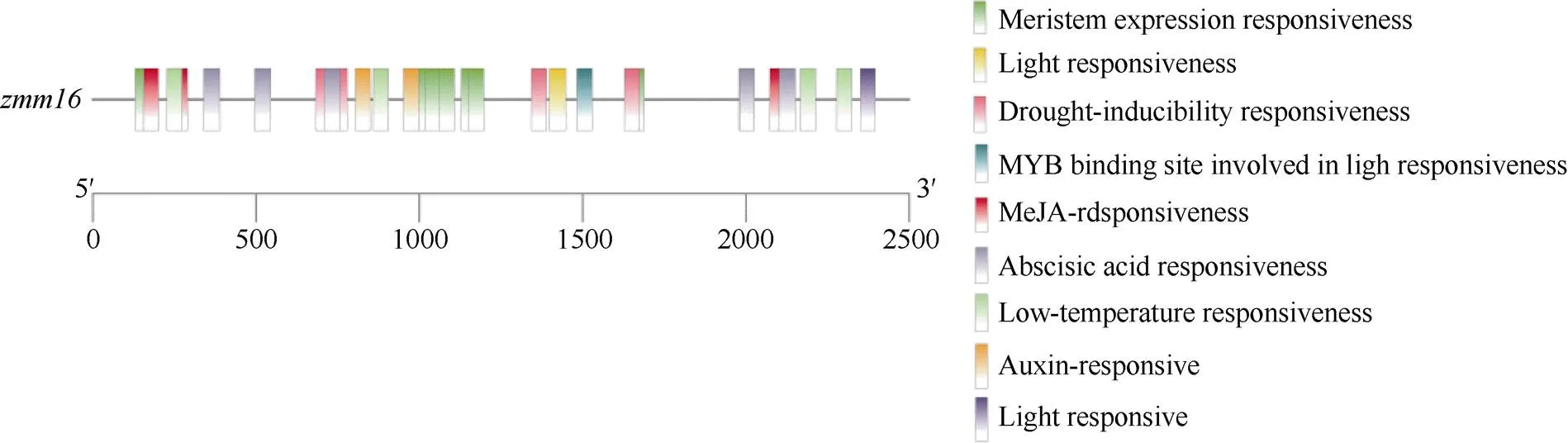
图4 zmm16启动子元件预测分析
2.5 zmm16表达量分析
分别取突变体和野生型幼穗各3株提取RNA, 进行RT-PCR。结果表明,中基因表达量降低。这说明转座子的插入突变并非导致完全不表达, 这一结果也对应了突变体中花药未完全退化的表型(图5)。
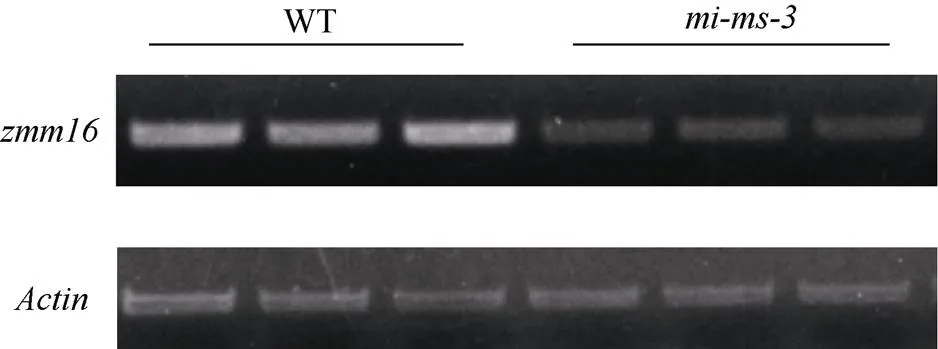
图5 zmm16表达量分析
相对于野生型,中基因表达量降低。
The expression ofindecreased compared with wild type.
3 讨论
与模式植物拟南芥相比, 玉米基因组更为复杂, 是拟南芥的几十倍。BSA法能够快速高效获得与目的基因连锁的分子标记, 发掘更多控制作物农艺性状的遗传位点。Zhao等[11]发现了一个玉米雌穗无花丝突变体, 利用BSA法筛选到与目的基因连锁的2个SSR标记umc1555和umc1448, 并通过精细定位及序列分析, 确定目的基因为。Sun等[12]通过BSA与测序技术相结合的方法定位到水稻耐冷性相关基因的候选区间, 并进一步通过GO分析确定了的候选基因。当转座子插入到功能基因内部或基因附近后能够破坏基因的功能从而产生突变表型。转座子均具有约220 bp保守末端反向重复序列[13], 因此,可作为插入标签用以进行基因定位。根据TIRs序列设计引物, 当转座子插入到某一基因后产生插入突变, 通过突变表型的筛选以及TIRs相关引物, 分离得到插入位点侧翼序列, 进而获得基因序列。Wang等[14]利用定位区间内25个候选基因的特异性引物与TIR引物组合扩增的方法, 成功克隆到一个雄性不育基因, 它编码一个半乳糖基转移酶, 主要在花药表皮和绒毡层中表达。Cui等[15]通过基因特异性引物和TIR引物组合扩增及测序的方法, 鉴定了四个等位基因插入的转座子的种类及其插入位点。本研究采用BSA结合转座子标签法的定位策略, 成功定位到突变基因。BSA能够快速找到与目的基因连锁的分子标记, 使从整个基因组上定位突变基因更为快速。相较于通过测序来确定突变基因的方法, 转座子标签法的利用可以直接通过PCR确定目的基因, 降低了试验成本。
Bartlett等[5]利用BSA法将基因定位到3号染色体长臂上, 并发现突变表型与umc1266完全连锁, 根据突变体表型推定该基因属于B类基因, 在umc1266标记3.39 Mb处发现1个B类基因, 测序发现突变体第3内含子上G突变为A,突变体第4内含子上G突变为A, 导致转录本不能发生正常剪接, 影响基因转录过程, 造成雄花不育, 花药完全退化为膜状结构, 雌花可育, 花丝增多。本研究获得了一个新的等位突变体, 该突变体表型与相似, 但相对于花药完全退化,则表现为部分花药未完全退化, 雄穗上部分小穗含有花药, 与野生型植株花药不同之处在于的花药只有2个药室, 同时雄穗上其他部分花药退化为膜状并在其末端形成丝状物。在突变体中,内含子发生点突变, 转录本不能进行正常剪接, 产生不完整的转录本, 导致基因功能缺失。而突变表型是由转座子的插入突变造成的。取雄穗进行表达量分析发现,突变体基因的表达量下降而非完全不表达, 这一突变表现为渗漏突变, 即基因编码的蛋白功能不完全失活, 仍然保留了一些功能。与相同之处在于,不育基因也与雌穗多花丝的标记性状[16]连锁,在开花期也表现为雌穗花丝增多, 每个子房对应2~4个花丝, 成熟果穗籽粒基部有败育种子。
高等植物的基因转录起始位点一般位于起始密码子(ATG)上游-40~-70 bp处。一般而言, 转录起始位点的缺失会影响转录, 但是也有学者认为转录位点并不是转录发生所必须的[17]。植物启动子中含有许多顺式作用元件, 通过转录因子与其结合来调控基因表达, 进而影响植物的生长发育过程。研究表明, MADS-box蛋白能与基因启动子区域中的顺式作用元件特异性结合, 使靶基因在特定的时间和空间以特定的强度表达[18]。中插入到基因上游, 可能导致基因编码蛋白不能与启动子区顺式作用元件结合来调控基因的表达, 而以一种低表达量的状态体现在突变体雄穗中部分小穗含有花药的表型上。基因启动子序列中存在低温响应元件及大量光响应元件, 这说明其表达可能受温度和光照环境因素的影响。在花药发育的特定阶段, 通过调节基因表达来适应一定范围内的光、温等环境变化。对玉米基因启动子区进行初步分析, 为将来进行基因功能的验证奠定了基础。
[1] 马冲, 张春庆, 陈举林, 侯玮, 王国胜. 玉米胞质雄性不育系研究进展. 中国农学通报, 2005, 21(1): 163–164. Ma C, Zhang C Q, Chen J L, Hou W, Wang G S. Advance on study of male sterility in maize., 2005, 21(1): 163–164 (in Chinese with English abstract).
[2] Tanaka W, Pautler M, Jackson D, Hirano H Y. Grass meristems II: inflorescence architecture, flower development and meristem fate., 2013, 54: 313–324.
[3] Li Q L, Liu B S. Genetic regulation of maize flower development and sex determination., 2017, 245: 1–14.
[4] Ambrose B A, Lerner D R, Ciceri P, Padilla C M, Yanofsky M F, Schmidt R J. Molecular and genetic analyses of thegene reveal conservation in floral organ specification between eudicots and monocots., 2000, 5: 569–579.
[5] Bartlett M E, Williams S K, Taylor Z, DeBlasio S, Goldshmidt A, Hall D H, Schmidt R J, Jackson D P, Whipple C J. The maize PI/GLO ortholog zmm16/sterile tassel silky ear1 interacts with the zygomorphy and sex determination pathways in flower development., 2015, 27: 3081–3098.
[6] 王关林, 方宏筠. 植物基因工程. 北京: 科学出版社, 2002. pp 742–744. Wang G L, Fang H J. Plant Genetic Engineering. Beijing: Science Press, 2002. pp 742–744 (in Chinese).
[7] Settles A M, Latshaw S, McCarty D R. Molecular analysis of high-copy insertion sites in maize., 2004, 32: e54.
[8] Chen C, Chen H, Yi Z, He Y, Hannah T R, Frank M H, Xia R. Tbtools: an integrative toolkit developed for interactive analyses of big biological data., 2020, 13: 1194–1202.
[9] Acosta I F, Helene L, Romero S P, Eric S, Mats H, Mottinger J P, Moreno M A, Dellaporta S L. Tasselseed1 is a lipoxygenase affecting jasmonic acid signaling in sex determination of maize., 2009, 323: 262–265.
[10] Chuck G. Molecular mechanisms of sex determination in monoecious and dioecious plants., 2010, 54: 53–83.
[11] Zhao Y, Zhang Y Z, Wang L J, Wang X R, Xu W, Gao X Y, Liu B S. Mapping and functional analysis of a maize silkless mutant., 2018, 9: 1127.
[12] Sun J, Yang L, Wang J, Liu H L, Zheng H L, Xie D W, Zhang M H, Feng M F, Jia Y, Zhao H W, Zou D T. Identification of a cold-tolerant locus in rice (L.) using bulked segregant analysis with a next-generation sequencing strategy., 2018, 11: 24.
[13] Bennetzen J L, Springer P S, Cresse A D, Hendrickx M. Specificity and regulation of the mutator transposable element system in maize., 1993, 12: 57–95.
[14] Wang D X, Skibbe D S, Walbot V. Maize(), a putative b-1,3-galactosyltransferase, modulates cell division, expansion, and differentiation during early maize anther development., 2013, 26: 329–338.
[15] Cui X Q, Hsia A P, Liu F, Ashlock D A, Wise R P, Schnable P S. Alternative transcription initiation sites and polyadenylation sites are recruited duringsuppression at thelocus of maize., 2003, 163: 685–698.
[16] 林晓怡, 杨典洱, 林建兴. 带遗传标记的玉米基因雄性不育的发现及遗传和利用研究. 作物学报, 2000, 26: 129–133.Lin X Y, Yang D E, Lin J X. The discovery, inheritance and utilization of genic male sterility with genetic marker in maize., 2000, 26: 129–133 (in Chinese with English abstract).
[17] 王颖, 麦维军, 梁成邺, 张明永. 高等植物启动子的研究进展. 西北植物学报, 2003, 23: 2040–2048. Wang Y, Mai W J, Liang C Y, Zhang M Y. Advances on studies of plant promoters., 2003, 23: 2040–2048 (in Chinese with English abstract).
[18] Becker A, Theissen G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants., 2003, 29: 464–489.
Genetic analysis and characterization of male sterile mutantin maize
TIAN Shi-Ke, QIN Xin-Er, ZHANG Wen-Liang, DONG Xue, DAI Ming-Qiu, and YUE Bing*
National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University, Wuhan 430070, Hubei, China
Maize is one of the best crops in the utilization of heterosis. Male sterile lines are important germplasms for the hybrids production. A male sterile mutant namedwas obtained by screening in a mutator insertion library. The number of male anthers in tassel decreased and not exserted. There were few anthers with only two pollen sacs in the mutant tassels, and some of the anthers were degenerated to membranous and formed filaments at their ends. Although pollens in the anthers could be stained by I2-KI, pollen shedding was abnormal and the number of pollen grains decreased. The number of silks in the ear of the mutant increased, and there was a sterile grain on both sides of the maturated kernel. Fertility of F1plants, which were obtained by hybridization betweenand maize inbred Mo17, was normal. Genetic analysis of F2population showed that the mutant phenotype was controlled by a recessive gene. The candidate gene was preliminarily mapped on the long arm of chromosome 3 by BSA and it was located between a SSR marker and an Indel marker with a distance of 1.5 cM. There are 21 candidate genes in this region. It was finally found that the insertion mutation oftransposon occurred at 30 bp upstream of the coding region of() by transponson tagging and sequencing analysis. The results showed thatwas a new allele of, which caused by a single base mutation in the coding region. RT-PCR analysis indicated that the expression ofin the mutant was decreased. The identification of the new allelic mutant ofin this study would provide new materials for the study of flower development and hybrid seed production.
maize; male sterile; gene mapping; genetic analysis
本研究由国家重点研发计划项目(2016YFD0100804)资助。
This study was supported by the National Key Research and Development Program of China (2016YFD0100804).
岳兵, E-mail: yuebing@mail.hzau.edu.cn
E-mail: tianshike1996@163.com
2020-05-07;
2020-08-19;
2020-08-31.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20200828.1751.009.html
10.3724/SP.J.1006.2020.03025
猜你喜欢
林业科学(2022年1期)2022-03-23 06:56:24
中国蜂业(2021年5期)2021-05-22 02:59:26
安徽医科大学学报(2016年12期)2017-01-15 14:21:44
浙江农林大学学报(2016年6期)2016-12-12 12:01:32
山东农业工程学院学报(2016年6期)2016-12-01 05:38:19
广西植物(2016年10期)2016-11-11 06:51:39
西南农业学报(2016年4期)2016-05-17 05:41:45
山东医药(2015年40期)2015-02-28 14:28:45
华东理工大学学报(自然科学版)(2014年5期)2014-02-27 13:49:27
食品科学(2013年23期)2013-03-11 18:30:11
