
基于BSA-Seq技术挖掘大豆中黄622的多小叶基因
2020-11-27 13:23:56张之昊刘章雄邱丽娟
作物学报 2020年12期
张之昊 王 俊 刘章雄 邱丽娟,*
基于BSA-Seq技术挖掘大豆中黄622的多小叶基因
张之昊1,2王 俊1刘章雄2,*邱丽娟1,2,*
1长江大学农学院, 湖北荆州 434025;2中国农业科学院作物科学研究所, 北京 100081
栽培大豆()叶片一般为三出复叶, 也有个别品种或植株突变体产生4~7个小叶, 为多小叶。复叶的形成使植物对外界环境的适应能力增强, 对大豆多小叶相关基因的挖掘和研究有助于改善大豆农艺性状和产量表现。本研究从大豆栽培品种中品661的突变体库中鉴定出一个多小叶突变体——中黄622, 每个复叶有4~9个小叶。利用该突变体与中品661配制组合, 分别于北京和海南调查F2和F2:3植株叶片表型, 结果表明, 多小叶性状受1对不完全显性基因控制。采用BSA-seq方法进行定位, 利用F2正常三出复叶和多小叶个体分别构建混池, 测序结果与参考基因组平均比对效率为98.83%, 平均覆盖深度为32.75´, 基因组覆盖度为99.22%。ED方法关联分析发现, 在11号染色体定位到2个区域, 总长度为5.29 Mb, 共包含1103个基因。根据SNP-index方法关联分析, 当置信度为0.99时, 在11号染色体鉴定出3个区域, 总长度为3.42 Mb, 共包含701个基因。2种关联分析方法同时定位的基因有690个, 亲本之间存在SNP的基因有6个。本研究结果为大豆多小叶基因图位克隆奠定了基础。
大豆; 突变体; 混池测序
叶片是植物进行光合作用和呼吸作用的重要器官, 对植物的生长发育起着重要作用。在植物界, 叶片的形态在不同物种间存在广泛的多样性, 单叶植物有一个与叶柄相连的叶片, 如水稻、玉米等, 而复叶植物的叶片由多个着生在叶轴上的小叶组成, 如番茄等。与单叶相比, 复叶虽然在叶片总面积上有所减少, 但其遭受外界的机械压力(如风、雨等)比单叶要小的多, 因此复叶的形成使植物更能适应恶劣的环境[1]。相关研究表明, 大豆多小叶单株全生育期不摘叶处理的部分农艺性状和产量表现均明显优于全生育期摘除多片叶的对照处理[2]。因此, 大豆多小叶相关基因的挖掘和研究有助于改善大豆农艺性状和产量表现。
大豆多小叶的遗传研究可追溯到1972年, Fehr[3]利用七小叶和五小叶大豆材料, 与三出复叶大豆进行两两杂交构建F2群体, 遗传分析表明, 五小叶和七小叶分别受显性基因和隐性基因控制。Jeong等[4]通过构建遗传图谱精细定位了控制五小叶的基因, 该基因编码一个假定AP2功能域, 但并没有对该基因进行进一步的功能验证。Devine[5]发现了控制七小叶的隐性基因与控制茸毛密度的基因存在连锁关系, Seversike等[6]鉴定出与基因连锁的分子标记, 从而将定位在B1连锁群(11号染色体)。傅来卿[7]通过对栽培大豆品种Wilkine进行物理诱变得到多小叶突变体, 该多小叶突变体每个复叶有4到7片小叶, 对该突变体进行遗传分析, 命名了一个控制多小叶的基因; 王克晶等[8]对五小叶的野生大豆进行遗传研究, 命名了基因和。总之, 大豆多小叶的相关研究多处于遗传分析和初定位阶段, 通过正向遗传学挖掘多小叶基因的相关研究鲜见报道。
突变体是进行正向遗传学研究的优异材料, 对随机诱变产生的突变体进行研究, 不仅能够挖掘新的基因, 也可以揭示已知基因的新功能[9-12]。随着新一代测序技术的发展, 基于BSA-Seq的方法挖掘新基因在作物农艺性状相关基因的定位研究中的应用日益广泛[12-16], 该方法在不构建遗传图谱的情况下高效快速地挖掘目的基因。本研究在栽培大豆中品661的突变体库中鉴定出一个多小叶突变体——中黄622, 该突变体在田间达到5个到9个小叶, 是研究大豆小叶数量的优异资源。利用BSA-Seq方法进行定位, 确定了6个候选基因, 为大豆多小叶基因图位克隆奠定了基础。
1 材料与方法
1.1 群体构建与表型鉴定
本试验通过EMS诱变, 并在田间筛选, 在栽培大豆中品661突变体库中鉴定出一个多小叶的不完全显性突变体中黄622, 2016年将中品661与中黄622进行杂交, 得到F0, 2017年6月将F1种植于北京顺义, 并于同年10月收获。2018年6月在顺义种植F2群体, 并于盛花期观察239个F2植株表型, 记录单株每个复叶的小叶数量并做记录。根据多小叶复叶的数目, 将单株分为3个等级: I级为多小叶复叶极多, 整个单株上仅有极少数(0~4个)叶片为正常三出复叶, 其余均为多小叶, 与突变体中黄622的表型相同; II级为多小叶复叶数目介于突变体中黄622和野生型之间, 至少有1个多小叶复叶; III级为整个单株所有叶片均为正常叶片, 与野生型相同。2018年10月收获F2单株的种子, 每个F2单株选取10粒以上的种子于2018年11月在海南种植F2:3群体, 于盛花期观察单株中每个复叶的小叶数量并做记录。通过观察F2在F2:3的分离情况, 来确定每个F2单株表型的准确性和稳定性。
1.2 混池测序
根据2个亲本及F2叶片表型, 构建2个亲本池和2个极端表型混池, 亲本池分别为10株中品661与10株突变体中黄622; 在F2植株中, 选择单株所有叶片均为三出复叶的30株个体构建正常叶片混池, 单株复叶均为多小叶30株个体构建多小叶混池。具体操作步骤如下: 首先, 在植株盛花期取植株顶端幼叶, 利用CTAB法分别提取单株叶片 DNA; 然后检测DNA浓度, 将不同池的植株DNA等量混合构建出4个混池。送北京百迈克生物科技有限公司测序。利用Illunima Casava 1.8进行碱基识别分析, 采用双端150 bp测序策略进行基因组测序。亲本池测序深度为10´, 后代混池测序深度为30´。参考基因组为Wm82.a2.v1版本的大豆基因组[17]。使用GATK[18]软件工具来实现SNP的检测, 利用SnpEff[19]软件进行变异注释和预测变异影响。
1.3 数据分析
1.3.1 关联分析 在关联分析前, 首先对SNP进行过滤, 过滤标准如下: 首先过滤掉有多个基因型的SNP位点, 其次过滤掉read支持度小于4的SNP位点, 最终得到高质量的可信SNP位点。
欧式距离(euclidean distance, ED)算法, 是利用测序数据寻找混池间存在显著差异标记, 并以此评估与性状关联区域的方法[20]。理论上, 多小叶混池和正常叶混池之间除了小叶数目性状相关位点存在差异, 其他位点均趋向于一致, 因此非目标位点的ED值应趋向于0。ED方法的计算公式如下所示, ED值越大, 表明该标记在多小叶混池和正常叶混池之间的差异越大。
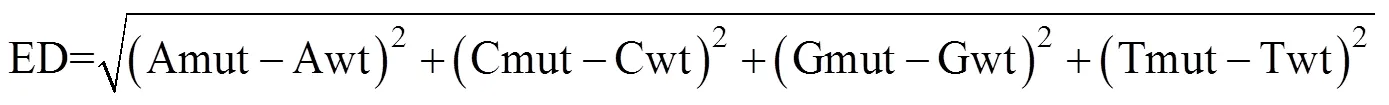
式中, Amut表示A碱基在突变混池中的频率, Awt表示A碱基在野生型混池中的频率; Cmut表示C碱基在突变混池中的频率, Cwt表示C碱基在野生型混池中的频率; Gmut表示G碱基在突变混池中的频率, Gwt表示G碱基在野生型混池中的频率; Tmut表示T碱基在突变混池中的频率, Twt表示T碱基在野生型混池中的频率。
在进行分析时, 利用多小叶混池和正常叶混池间基因型存在差异的SNP位点, 统计各个碱基在2个混池中的深度, 并计算每个位点ED值, 为消除背景噪音, 本试验采用原始ED的5次方作为关联值以达到消除背景噪音的功能[20]。
SNP-index是一种通过混池间的基因型频率差异进行标记关联分析的方法[20-21], 主要是寻找混池之间基因型频率的显著差异, 用Δ(SNP-index)统计。标记SNP与性状关联度越强, Δ(SNP-index)越接近于1。Δ(SNP-index)计算公式如下:
SNP-index (Mut) = ρx/(ρX + ρx)
SNP-index (WT) = ρx/(ρX + ρx)
ΔSNP-index=SNP-index(Mut) − SNP-index(WT)
式中, Mut为子代的突变池, 即多小叶混池, WT代表子代的野生池即正常叶混池, ρX和ρx分别为野生型亲本中品661的等位基因和突变型亲本中黄622的等位基因在各自池中出现的read数目。通过ΔSNP-index 可以观察每个位点在多小叶混池和正常叶混池之间的差异。
为了消除假阳性的位点, 利用标记在基因组上的位置, 可对同一条染色体上标记的ΔSNP-index值进行拟合[21], 本研究采用DISTANCE方法对ΔSNP-index进行拟合, 然后根据关联阈值, 选择阈值以上的区域作为与性状相关的区域。
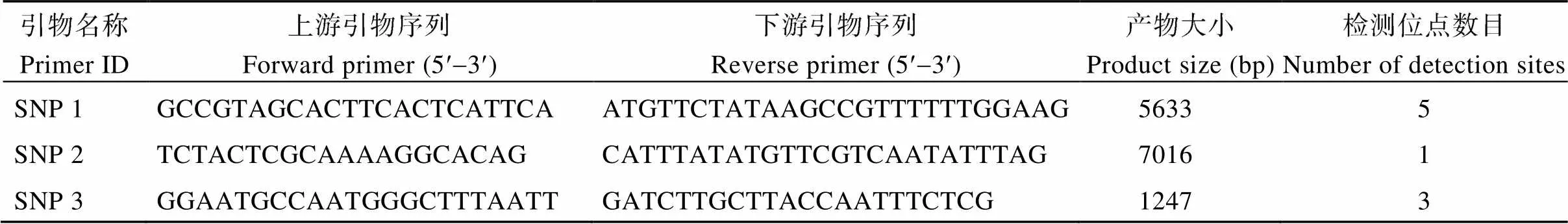
1.3.2 变异位点的鉴定 为了检测过滤SNP方法的可靠性, 选取了1个高质量的SNP位点和其附近的8个低质量的SNP位点进行验证。首先根据比对参考基因组, 在SoyBase (http://www.soybase.org/)基因组数据库中利用BLAST的方法在变异位点上下游查找20 bp左右的特异序列作为扩增变异位点的引物, 并通过DNAMAN软件(https://www.lynnon. com/)预测引物退火温度和计算GC%值。在保证引物特异性的前提下, 调整引物序列长度, 使退火温度在55~65℃, GC%值在40%到60%之间。同一对引物的上下游序列的退火温度不超过3℃。利用PCR扩增目的片段, 对PCR产物的目标位点进行测序, 鉴定该位点是否存在。
1.3.3 候选区域基因的功能注释 应用BLAST[22]软件对候选区间内的编码基因进行多个数据库(NR[23]、Swiss-Prot、GO[24]、KEGG[25]、COG[26])的深度注释。本研究的BSA-Seq数据分析流程图如图1所示。
1.3.4 数据可视化 本研究利用circos软件(http://circos.ca/)对重测序分析得到的结果进行作图, 使测序分析结果和关联分析结果可视化。
1.4 候选基因表达分析
为了研究关联区域内候选基因在不同组织中的表达情况, 利用Phytozome(http://phytozome.jgi. doe.gov/)的基因表达数据库筛选6个基因的表达, 研究的表达部位依次为: 茎端分生组织、叶片、花、荚、茎、节、根和根毛。利用R3.6.2 (http://www.r-project. org/)将表达数据进行标准化并制作基因表达图谱。
2 结果与分析
2.1 多小叶遗传分析
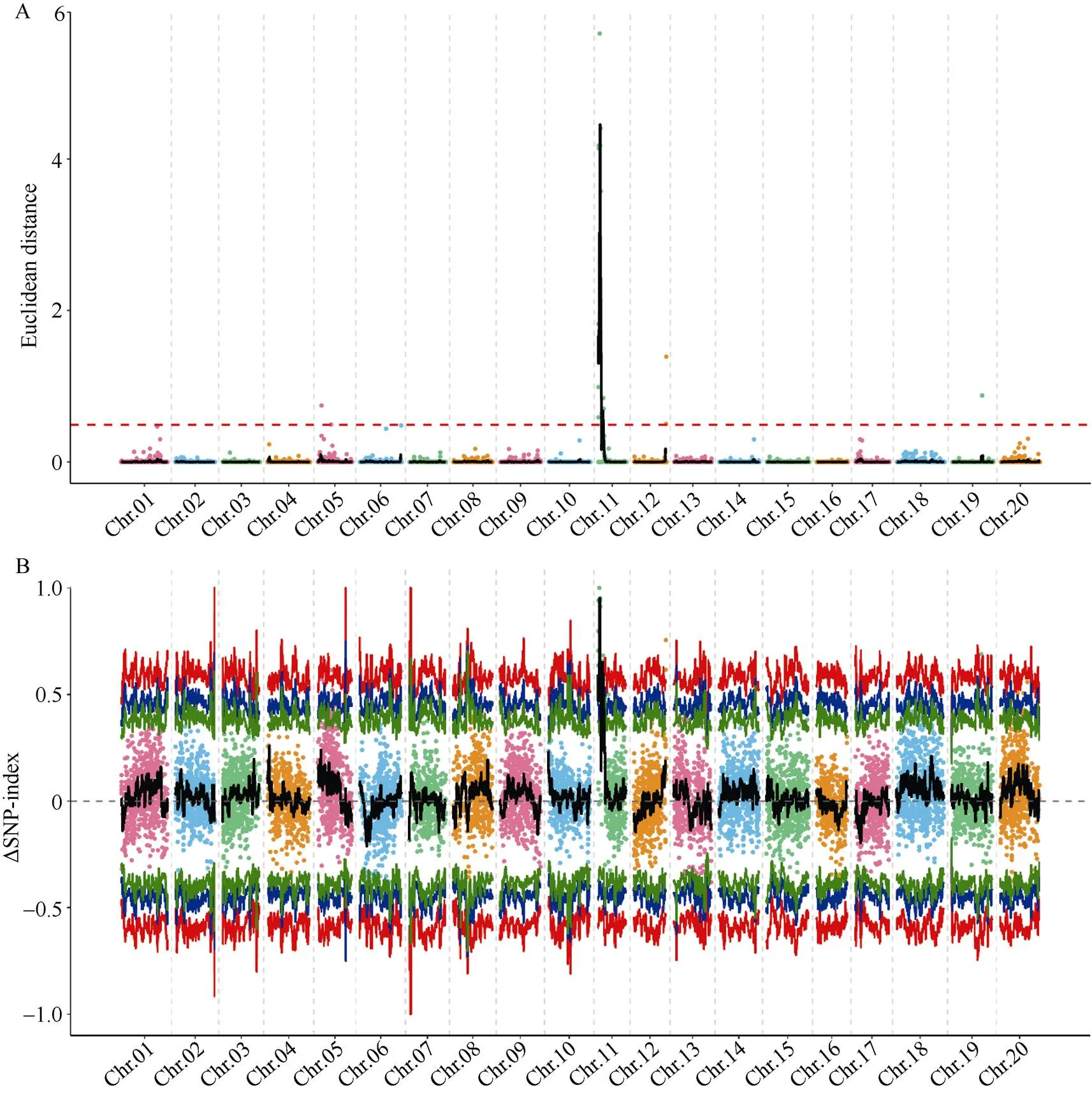
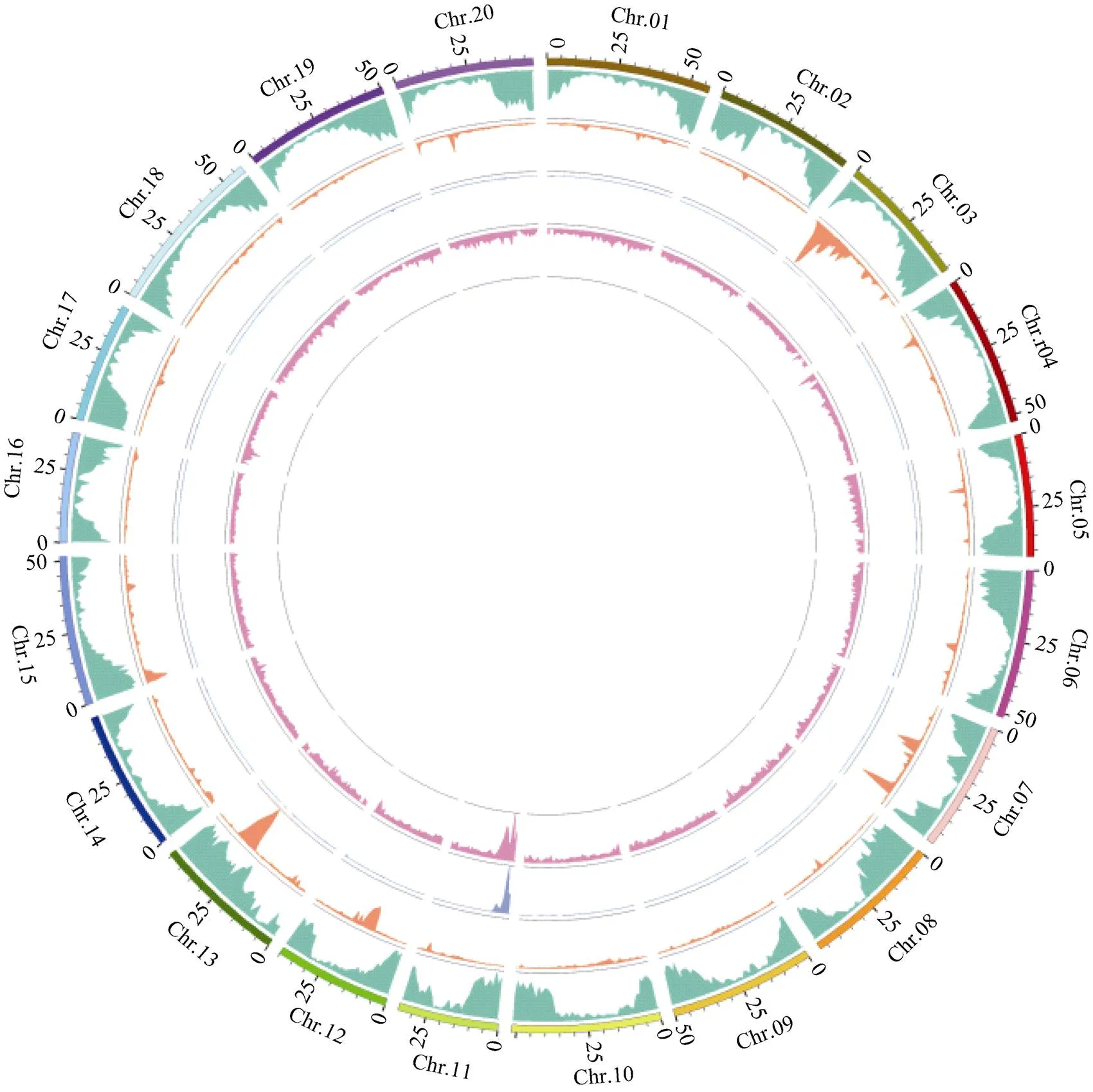
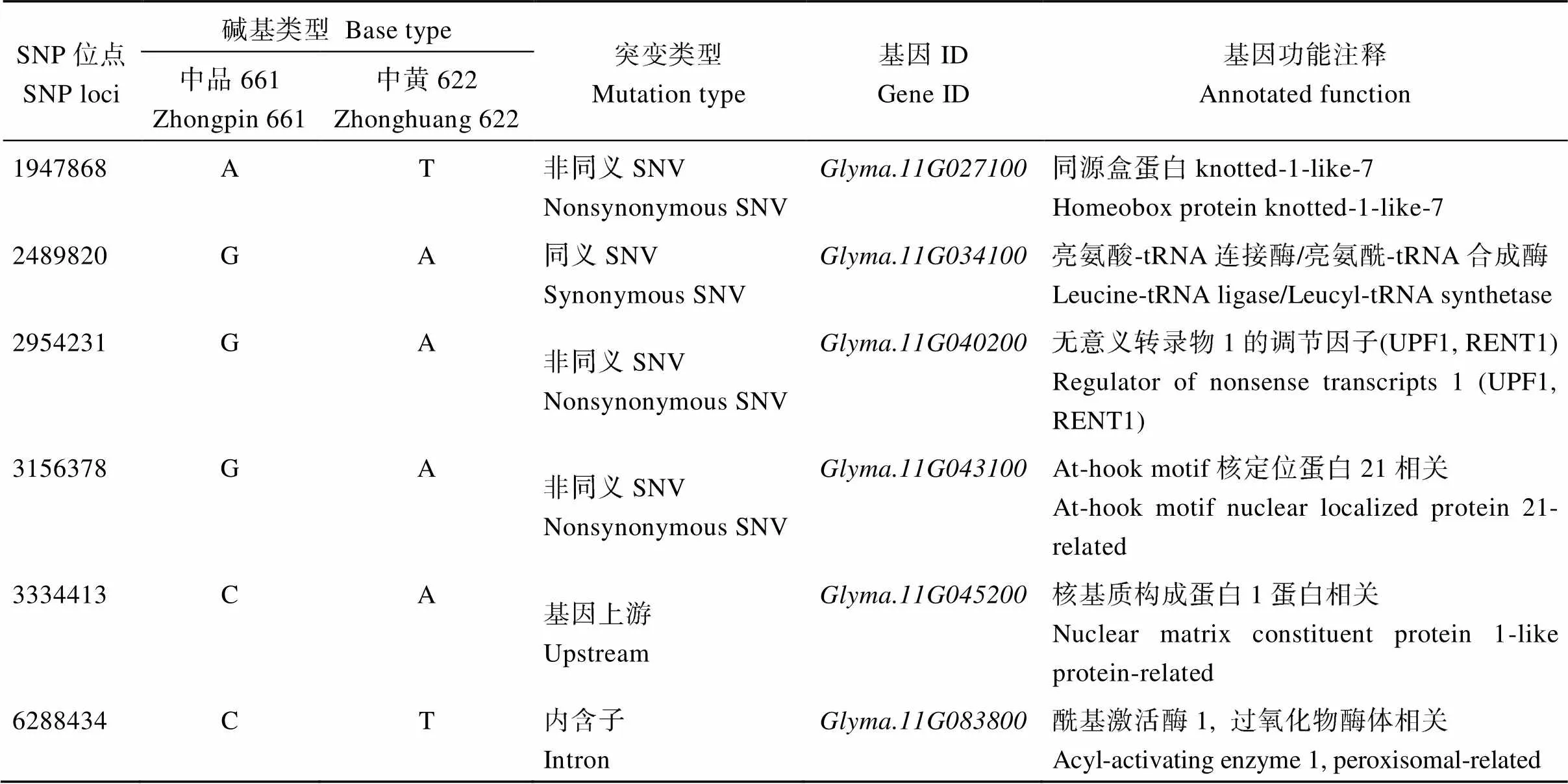
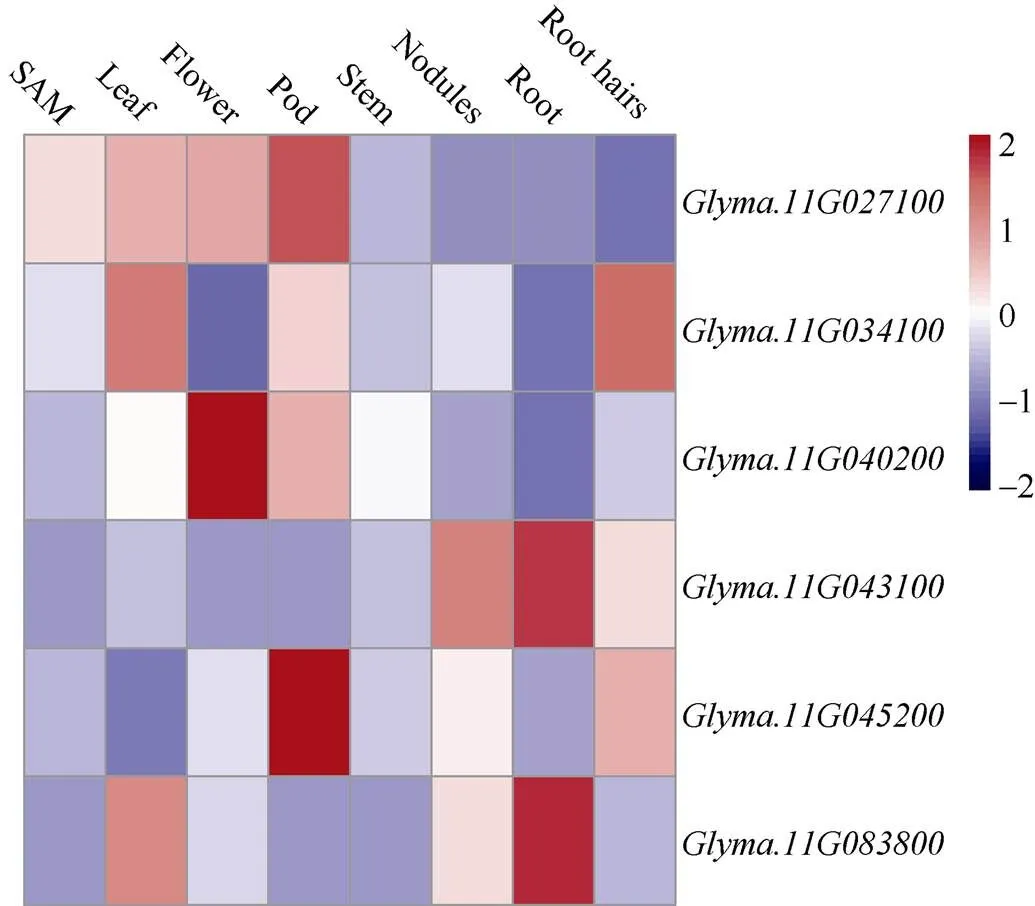
突变体中黄622与野生型中品661相比, 叶片多为5到9片的多小叶复叶, 但也有少量单株出现0~4片正常的三出复叶。通过观察F2:3的表型及分离情况, 确定对应F2单株的基因型。观察发现, 多小叶等位基因纯合基因型(lf lf )单株的表型均为I型, 正常叶等位基因纯合基因型(lf lf )单株的表型均为III型, 而杂合单株(lf lf )的表型绝大多数为II型, 仅有极少量的I型(2个)和III型(4个)(表1), 这说明杂合单株的表型不稳定。表明, 该多小叶性状受1对不完全显性基因控制, 且杂合基因型单株的表型可能受到环境和遗传背景的影响, 表现出不同程度的多小叶性状。卡方检测结果表明, 3种基因型的F2单株数目不符合1∶2∶1 (c20.05= 5.99 图1 BSA-Seq数据分析流程 表1 F2群体中不同基因型和表型的单株数目 I~III: 多小叶性状不同等级的表型。I: 与突变体表型相同; II: 表型介于突变体和野生型之间; III: 与野生型相同。lf lf 、lf lf 、lf lf : 利用F2:3的分离情况鉴定的F2不同的基因型的单株。lf lf : 多小叶纯合基因型;lf lf : 正常叶纯合基因型;lf lf : 杂合基因型。 I–III: leaflet number traits of different degree of phenotypes. I: the same as the mutant surface type; II: the phenotype is between the mutant and the wild type; III: the same as the wild type. lf lf ,lf lf ,lf lf : different genotypes of F2individuals.lf lf : homozygous genotype of multifoliolate leaf;lflf: homozygous genotype of trifoliate leaves;lflf : heterozygous genotype. 2.2.1 数据质控 对从F2选择30个多小叶单株和30个正常叶单株分别构建混合池, 与2个亲本通过Illumina HiSeq重测序, 过滤后得到的碱基数目为144.84 Gbp, Q30>91.61%, GC含量在35.81%~ 36.94%, 插入片段大小的分布呈单峰的正态分布, 样品与参考基因组平均比对效率为98.83%, 平均覆盖深度为 32.75´, 基因组覆盖度为99.22% (至少1个碱基覆盖)。这些参数说明, 测序数据质量合格, 与大豆参考基因组比对效率较高, 可用于后续变异检测及性状的基因定位。 2.2.2 关联分析 进一步过滤SNP, 得到高质量可信SNP位点12,160个。采用ED算法和SNP-index 2种算法对这些高质量SNP进行关联。 利用ED方法计算关联值, 并取原始ED的5次方作为关联值以达到消除背景噪音的功能, 然后采用DISTANCE方法对ED值进行拟合。取所有位点拟合值的median+3SD作为分析的关联阈值[20], 计算得0.49。根据关联阈值判定, ED关联结果如图2-A所示, 在11号染色体共得到2个区域, 总长度为5.29 Mb, 共包含1103个基因(表2)。 2个混池的ΔSNP-index的分布如图2-B所示。当置信度为0.99时, 利用SNP-index方法共关联到3个区域, 总长度为3.42 Mb, 共包含701个基因(表2)。2种关联分析方法得到的结果取交集, 共得到3个区域(表2), 总长度3.36 Mb, 共包含690个基因。结合上述内容, 将样品的变异结果及BSA关联分析结果使用circos软件(http://circos.ca/)作图(图3), 将测序分析结果和关联分析结果可视化。 2.2.3 候选基因的筛选 根据原始测序数据, 区间内的SNP有43个, 其中高质量的SNP有6个, 低质量的SNP有37个。为了验证过滤SNP方法的可靠性, 进一步筛选候选基因, 本试验设计了3对引物(表3), 分别用以鉴定1个高质量的SNP及其2侧的低质量SNP位点。其中引物SNP2的扩增产物片段全长7016 bp, 包含1个高质量的SNP位点, 而引物SNP1扩增产物片段全长5633 bp, 包含该高质量SNP上游的5个低质量位点, 引物SNP3扩增产物片段全长1247 bp, 包含该高质量SNP下游的3个低质量位点。对两亲本PCR产物进行测序鉴定。两亲本序列比对结果表明, 中品661和中黄622在8个低质量的SNP位点处均无变异, 而2个亲本在高质量SNP处的测序结果与重测序结果一致(表4)。这些结果为候选基因的筛选提供了参考依据。 图2 利用2种关联方法鉴定多小叶基因候选区间 A: ED关联分析结果, 横坐标为染色体位置, 纵坐标代表拟合后的欧式距离(ED)值的5次方, 黑色的线为拟合后的ED的5次方作关联值, 红色的虚线代表显著性关联阈值。B: SNP-index关联分析结果, 横坐标为染色体位置, 黑色的线为拟合后的ΔSNP-index值, 红色的线代表置信度为0.99的阈值线, 蓝色的线代表置信度为0.95的阈值线, 绿色的线代表置信度为0.90的阈值线。2种关联分析结果均表明, 与多小叶相关的关联区域位于11号染色体末端。 A: ED correlation analysis results, the abscissa is the chromosome position, the ordinate represents the fifth power of the euclidean distance (ED) value after fitting, the black line is the fifth power of ED after fitting, the dashed line represents the significance association threshold. B: SNP-index correlation analysis results, the abscissa for chromosomal location, the black line for fitting after ΔSNP-index value, the red line represents the confidence level of 0.99 the threshold line, blue line represents the confidence level of 0.95 the threshold line, green line represents the confidence level of the threshold line of 0.90. The results of the two association analysis methods show that the correlation regions associated with the multifoliolate leaf trait is located at the end of chromosome 11. 表2 利用不同方法在11号染色体获得关联区域 图3 样品间SNP及关联信号在染色体上的分布 从外到里依次为: 第1圈: 染色体坐标; 第2圈: 基因分布; 第3圈: SNP密度分布; 第4圈: ED值分布; 第5圈: ΔSNP-index值分布。 From outside to inside in order: the first circle represents chromosome coordinates, the second circle represents gene distribution, the third circle represents SNP density distribution, the fourth circle represents ED value distribution, and the fifth circle represents ΔSNP-index value distribution. 表3 引物序列和信息 表4 对区间内部分SNP位点进行鉴定 混池read值中, 逗号前为SNP位点参考碱基的read值, 逗号后为突变碱基的read值。 Among read values in the mixed pool, the number before the comma is the read value of the SNP loci reference base, and the number after the comma is the read value of the altered base. 根据不同质量SNP的鉴定结果, 本研究对区间内的候选基因进行了进一步的筛选。筛选到定位区间内亲本间存在高质量的SNP的6个基因上(表5)。在这6个基因中, 有3个基因发生了非同义突变, 导致氨基酸序列发生变异, 1个基因发生了同义突变, 其余2个基因的突变分别发生在基因上游和内含子上, 该结果为多小叶候选基因的发掘提供了信息。 2.2.4 候选基因表达的生物信息学分析 为了进一步探究共定位区间内候选基因在各个组织中的表达情况, 利用phytozome (phytozome.jgi.doe. gov/)数据库查询了6个基因在Williams 82中的表达数据, 从构建的表达图谱(图4)可以看出,的表达谱较广, 在茎端分生组织、叶、花和荚中均有较高表达;和在叶片中的表达都比较高, 但在分生组织中表达量较低, 其中在节和根中也有较高的表达量, 而则在根毛中高表达;在叶和茎中的表达量居中, 在花和荚中有着较高的表达, 在茎端分生组织和其他部位表达较低;在节、根、根毛表达较高, 在其他组织中表达较低;在荚、节和根毛中表达较高, 在其他部位表达较低。考虑到相关基因在分生组织和叶片中的表达以及表达量的变化对复叶发育的重要性,推测在分生组织和叶片中表达较高的可能与复叶发育相关的基因, 在分生组织中表达较低, 但在叶片中表达较高的和也有可能是复叶发育相关的基因。但候选基因在中品661与中黄622中是否与Williams 82的表达模式一致, 仍需要后期验证。 表5 2种方法共定位区间内亲本之间SNP类型 图4 6个候选基因的表达图谱 方块内颜色显示候选基因表达水平: 蓝色最低, 白色居中, 红色最高。 Colors in the square represent the expression level of candidate genes: blue is the lowest, white is middle, and red is the highest. 与复叶发育相关的基因包括、、等[27]。(KNOTTEDlike homeobox)基因家族被认为是茎端分生组织(SAM)分化出叶原基和维持形态建成的关键基因, 叶原基形成初期(P0),的表达会提高, 但在叶原基形成之后表达降低[28-29], 在单叶植物如玉米()中, 这种低表达是永久性的[30], 而在复叶植物如番茄和碎米荠(L.)中,基因的表达会再次升高[29,31]。在转基因的拟南芥中,基因家族中任何一个基因的异位表达都会在引起叶边缘产生许多浅裂形成的小叶[32]。研究发现,基因与复叶植物的形态建成有明显的关系, 在基因参与复叶发育的物种中(如番茄), 如果增加基因的表达, 可以产生明显的复叶重复结构单元[31,33]。基因家族是在被子植物叶发育起着关键作用的另外一类基因, 它和基因的相互拮抗作用共同决定叶的发生。基因的功能是维持分生组织属性, 而基因则参与侧生器官的发生和叶极性的建立等分化相关的发育过程[34-36]。另外, 相关研究表明,在豌豆和百脉根中的直系同源基因()和(), 参与控制小叶原基的起始和形态发生[27,37-38]。 关于大豆多小叶的基因定位研究较为深入的是和基因, 在前期报道中, Jeong等[39]定位到了控制大豆五小叶的基因, 该基因编码一个假定AP2功能域, 虽然并未对该基因进行功能验证, 但有研究表明AP2转录因子参与调控玉米叶片发育, 影响玉米叶形。这说明影响植物叶片发育的基因除了、等研究比较深入的基因外, 还有其他基因有待发掘和验证。 Seversike等[6]在不同群体中验证了基因与SSR标记Sat_272的连锁关系(LOD>4.0), 但并未得到定位区间。本研究通过ED和ΔSNP-index 2种方法进行关联分析, 将中黄622的多小叶相关基因定位在11号染色体末端, 经2种关联方法取交集后的关联区域大小3.36 Mb, 包含690个基因, 其中关联区域内部发生的SNP有6个, 分布在6个基因上。外显子发生SNP的基因有4个, 其中1个基因发生的变异为同义突变, 3个基因的变异导致氨基酸序列的变化。利用数据库(SoyBase)对比Sat_272 (物理位置为2,718,892至2,719,123)与本研究定位区间的物理位置发现, Sat_272在本研究的候选区间内。然而, 本研究定位的基因是否为多小叶基因还有待进一步研究验证。 Fehr[3]通过对三出复叶Hawkeye与七小叶突变体T255构建的群体的遗传分析发现, 小叶数目性状受1对基因控制, 控制正常的三出复叶的等位基因表现为完全显性, 但对T255和五小叶突变体T143构建的群体进行遗传分析发现,等位基因并不是在所有遗传背景中都是完全显性的。Seversike等[6]在七小叶大豆PI 548232和3个正常三出复叶大豆(Trill、MN1401、MN1801)构建的群体中再次证明为控制七小叶的隐性基因, 而杂合单株整株为正常三出复叶或有一片复叶为四小叶。本研究中, 多小叶突变体的复叶有5到9个小叶, 控制多小叶性状的基因为1对不完全显性基因, 杂合单株表型介于突变体和野生型之间, 且表型并不稳定, 在105个F2杂合单株中有6个单株表现出与亲本相近的表型。这说明, 多小叶性状的表型不稳定, 特别是杂合单株在不同的环境和遗传背景下有着不同的表型。因此, 对多小叶全面的遗传分析需要在多年多点不同的材料之间进行。 本研究定位区间内有6个候选基因(、、、、、)发生了SNP变异。是II类亚家族()成员, 该基因在茎端分生组织(SAM)和叶片中均有着较高的表达。虽然在植物复叶发育过程中, I类亚家族()基因是SAM分化出叶原基和维持形态建成的关键基因, 但尚未有基因与叶片发育相关的报道。为一个编码亮氨酸-tRNA连接酶/亮氨酰-tRNA合成酶的基因, 其经典功能是催化合成亮氨酰-tRNA直接参与遗传信息的解码合成蛋白质[40]。编码的无义转录物1的调节因子()参与调控无义介导的mRNA降解途径, 该途径能够识别并降解包括前终止的无义mRNA在内的异常mRNA[41-42], 并避免异常mRNA翻译成潜在有毒蛋白质, 从而对机体产生毒害效应。该途径的调控对基因的准确表达和机体细胞正常的生理活动起着重要作用[43-45]。是一个AT-hook蛋白相关的基因, AT-hook蛋白可能在植物生长发育中起着重要作用, 相关研究表明, 拟南芥中基因在过量表达时会延迟拟南芥的开花[46];基因的产物与核基质构成蛋白1相关, 核基质蛋白不仅是组成细胞核内部结构的支架, 而且同DNA的复制、RNA 的合成、激素的联接、基因表达的调节密切相关[47]。为一个酰基激活酶1基因, 与过氧化氢酶体相关。这些基因对大豆复叶形成和发育的影响将在后续的工作中进行验证。 突变体中黄622的多小叶受1对不完全显性基因控制, 基于BSA-Seq技术将该基因定位在11号染色体上, 定位区间内亲本之间存在SNP的基因有6个, 为大豆多小叶基因图位克隆创造了条件。 [1] Vogel S. Leaves in the lowest and highest winds: temperature, force and shape., 2009, 183: 13–26. [2] 宗春美, 岳岩磊, 邵广忠, 童淑媛, 徐显利, 杜震宇, 任海祥. 多小叶源对大豆光合特性和产量的影响. 大豆科学, 2010, 29: 627–626. Zong C M, Yue Y L, Shao G Z, Tong S Y, Xu X L, Du Z Y, Ren H X. Effects of multifoliolate compound leaf on photosynthetic characteristics and yield of soybean., 2010, 29: 627–626 (in Chinese with English abstract). [3] Fehr W R. Genetic control of leaflet number in soybeans., 1972, 12: 221–224. [4] Jeong S C, Kim J H, Bae D N. Genetic analysis of thegene that controls leaflet number in soybean., 2017, 130: 1685–1692. [5] Devine T E. Theandloci define soybean linkage group 16., 2003, 43: 2028–2030. [6] Seversike T M, Ray J D, Shultz J L, Purcell L C. Soybean molecular linkage group B1 corresponds to classical linkage group 16 based on map location of thegene., 2008, 117: 143–147. [7] 傅来卿. 大豆双复叶和多小叶突变体的研究. 大豆科学, 1986, 5: 283–288. Fu L Q. Study of mutants with opposite trifoliate leaves and multi-leaflet leaves in soybean., 1986, 5: 283–288 (in Chinese with English abstract). [8] 王克晶, 李福山, 周涛, 许占有. 来源于野生大豆的多小叶性状遗传分析. 大豆科学, 2001, 20: 22–25. Wang K J, Li F S, Zhou T, Xu Z Y. Inheritance of a five leaflet character arising from wild soybean (Sieb. et Zucc.) in soybeans ((L.) Merr.)., 2001, 20: 22–25 (in English with Chinse abstract). [9] Nawy T, Bayer M, Mravec J, Friml J, Birnbaum K D, Lukowitz W. The GATA factoris required to position the proembryo boundary in the earlyembryo., 2010, 19: 103–113. [10] Gallavotti A, Long J A, Stanfield S, Yang X, Jackson D, Vollbrecht E, Schmidt R J. The control of axillary meristem fate in the maize ramosa pathway.(Cambridge, England), 2010, 137: 2849–2856. [11] Vlad D, Kierzkowski D, Rast M I, Vuolo F, Ioio R D, Galinha C, Gan X, Hajheidari M, Hay A, Smith R S, Huijser P, Bailey C D, Tsiantis M. Leaf shape evolution through duplication, regulatory diversification, and loss of a homeobox gene., 2014, 343: 780–783. [12] Stewart G C, Roeder A H K, Patrick S, Chris S, Wolfgang L, Hector C. A genetic screen for mutations affecting cell division in theembryo identifies seven loci required for cytokinesis., 2016, 11: e0146492. [13] Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M, Innan H, Cano L, Kamoun S, Terauchi R. Genome sequencing reveals agronomically important loci in rice using MutMap., 2012, 30: 174–178. [14] Abe A1, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M, Innan H, Cano L, Kamoun S, Terauchi R. QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations., 2013, 74: 174–183. [15] Zhang H, Wang X, Pan Q, Li P, Liu Y, Lu X, Zhong W, Li M, Han L, Li J, Wang P, Li D, Liu Y, Li Q, Yang F, Zhang Y M, Wang G, Li L. QTG-Seq accelerates QTL fine mapping through QTL partitioning and whole-genome sequencing of bulked segregant samples., 2019, 12: 426–437. [16] Klein H, Xiao Y, Conklin P A, Govindarajulu R, Kelly J A, Scanlon M J, Whipple C J, Bartlett M. Bulked-segregant analysis coupled to whole genome sequencing (BSA-Seq) for rapid gene cloning in maize., 2018, 8: 3583– 3592. [17] Song Q J, Jenkins J, Jia G F, Hyten D L, Pantalone V, Jackson S A. Construction of high resolution genetic linkage maps to improve the soybean genome sequence assembly Glyma1.01., 2016, 17: 33. [18] McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo M A, The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data., 2010, 20: 1297–1303. [19] Cingolani P, Platts A, Wang L L, Coon M, Nguyen T, Wang L, Land S J, Lu X Y, Ruden D M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome ofstrain1118;-2;-3., 2012, 6: 80–92. [20] Hill J T, Demarest B L, Bisgrove B W, Gorsi B, Su Y C, Yost H J. MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq., 2013, 23: 687–697. [21] Fekih R, Takagi H, Tamiru M, Abe A, Natsume S, Yaegashi H, Sharma S, Sharma S, Kanzaki H, Matsumura H, Saitoh H, Mitsuoka C, Utsushi H, Uemura A, Kanzaki E, Kosugi S, Yoshida K, Cano L, Kamoun S, Terauchi R. MutMap+: Genetic mapping and mutant identification without crossing in rice., 2013, 8: e68529. [22] Altschul S F, Madden T L, Schäffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs., 1997, 25: 3389–3402. [23] Deng Y, Li J Q, Wu S F, Zhu Y P, Chen Y W, He F C. Integrated nr database in protein annotation system and its localization., 2006, 32: 71–72. [24] Ashburner M, Ball C A, Blake J A, Botstein D, Butler H, Cherry M, Davis A P, Dolinski K, Dwight S S, Eppig J T, Harris M A, Hill D P, Issel-Tarver L, Kasarskis A, Lewis S, Matese J C, Richardson J E, Ringwald M, Rubin G M, Sherlock G. Gene ontology: tool for the unification of biology., 2000, 25: 25–29. [25] Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome., 2004, 32: D277–D280. [26] Tatusov R L, Galperin M Y, Natale D A, Koonin E V. The COG database: a tool for genome-scale analysis of protein functions and evolution., 2000, 28: 33–36. [27] 杨霞, 高金珊, 杨素欣. 豆科复叶发育分子遗传机制的研究进展. 植物生理学报, 2017, 53: 905–915. Yang X, Gao J S, Yang S X. Progress of molecular mechanism of compound leaf development in legume plants., 2017, 53: 905–915 (in Chinese with English abstract). [28] Long J A, Moan E I, Medford J I, Barton M K. A member of the KNOTTED class of homeodomain proteins encoded by thegene of.(London), 1996, 379: 66–69. [29] Bharathan G, Goliber T E, Moore C, Kessler S, Pham T, Sinha N R. Homologies in leaf form inferrfromgene expression during development., 2002, 296: 1858–1860. [30] Lincoln C, Long J, Yamaguchi J, Serikawa K, Hake S. A-like homeobox gene inis expressed in the vegetative meristem and dramatically alters leaf morphology when overexpressed in transgenic plants., 1994, 6: 1859–1876. [31] Hay A, Tsiantis M. The genetic basis for differences in leaf form betweenand its wild relative., 2006, 38: 942–947. [32] Shani E, Burko Y, Ben-Yaakov L, Berger Y, Amsellem Z, Goldshmidt A, Sharon E, Ori N. Stage-specific regulation ofleaf maturation by class 1 KNOTTED1- LIKE HOMEOBOX proteins., 2009, 21: 3078–3092. [33] Hareven D, Gutfinger T, Parnis A, Eshed Y, Lifschitz E. The making of a compound leaf: genetic manipulation of leaf architecture in tomato., 1996, 84: 735–744. [34] Byrne M E, Barley R, Curtis M, Arroyo J M, Dunham M, Hudson A, Martienssen R A.mediates leaf patterning and stem cell function in., 2000, 408: 967–971. [35] Waites R, Selvadurai H R N, Oliver I R, Hudson A. Thegene encodes a MYB transcription factor involved in growth and dorsoventrality of lateral organs in., 1998, 93: 779–789. [36] Kim M, Pham T, Hamidi A, McCormick S, Kuzoff R K, Sinha N. Reduced leaf complexity in tomato wiry mutants suggests a role forandgenes in generating compound leaves., 2003, 130: 4405–4415. [37] Taylor S, Hofer J, Murfet I., the pea ortholog ofand, is required for normal development of flowers, inflorescences, and leaves., 2001, 13: 31–46. [38] Dong Z C, Zhao Z, Liu C W, Luo J H, Yang J, Huang W H, Hu X H, Wang T L, Luo D. Floral patterning in., 2005, 137: 1272–1282. [39] Jiang F K, Guo M, Yang F, Duncan K, Jackson D, Rafalski A, Wang S C, Li B L. Mutations in an AP2 transcription factor- like gene affect internode length and leaf shape in maize., 2012, 7: e37040. [40] Soll M D, Ibba T M. Aminoacyl-tRNA synthesis., 2000, 69: 617–650. [41] Fatscher T, Boehm V, Gehring N H. Mechanism, factors, and physiological role of nonsense-mediated mRNA decay., 2015, 72: 4523–4544. [42] 柴宝峰, 王美, 石文鑫, 柴杨丽, 吕佳. 无义mRNA降解途径的机制与进化.山西大学学报(自然科学版), 2017, 40: 639–644. Chai B F, Wang M, Shi W X, Chai Y L, Lyu J. Mechanism and evolution of nonsense-mediated mRNA decay.(Nat Sci Edn), 2017, 40: 639–644 (in Chinese with English abstract). [43] 贾晓波, 胡剑. 无义介导的mRNA降解. 中国生物化学与分子生物学报, 2012, 28(2): 22–27. Jia X B, Hu J. Nonsense-mediated mRNA decay., 2012, 28(2): 22–27 (in Chinese with English abstract). [44] Yamashita A. Role of SMG-1-mediated Upf1 phosphorylation in mammalian nonsense-mediated mRNA decay., 2013, 18: 161–175. [45] Bhattacharya A, Köhrer C, Mandal D, Rajbhandary U L. Nonsense suppression in archaea., 2015, 112: 6015–6020. [46] 肖朝文, 陈福禄, 傅永福.AT-hook基因过量表达延迟拟南芥开花. 中国农业科技导报, 2009, 11(4): 93–98. Xiao C W, Chen F L, Fu Y F. Over-expression of AT-hook genecan delay the flowering of., 2009, 11(4): 93–98 (in Chinese with English abstract). [47] Getzenberg R H, Pienta K J, Ward W S, Coffey D S. Nuclear structure and the three-dimensional organization of DNA., 1991, 47: 289–299. Mapping of an incomplete dominant gene controlling multifoliolate leaf by BSA-Seq in soybean (L.) ZHANG Zhi-Hao1,2, WANG Jun1, LIU Zhang-Xiong2,*, and QIU Li-Juan1,2,* 1School of Agriculture, Yangtze University, Jingzhou 434025, Hubei, China;2Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing 100081, China The leaves of cultivated soybean (L.) are comprising of three leaflets in general, but there are also individual varieties or mutants which have a high frequency of compound leaves with 4–7 leaflets, named multifoliolate leaves. Compound leaf formation enhances the plant's ability to adapt to the external environment. Study of related genes to multifoliolate leaves might contribute to the improvement yield level of and soybean agronomic traits. In this study, a multifoliolate leaf mutant Zhonghuang 622 was identified from the mutant library of soybean cultivar Zhongpin 661, which had 4–9 leaflets in each compound leaf. The compound leaf phenotypes of F2and F2:3population from a cross between Zhongpin 661 and Zhonghuang 622 were investigated in Beijing and Hainan, respectively. Analysis of phenotypic data from F2and F2:3population revealed that the multifoliolate leaf trait was controlled by an incomplete dominant gene. BSA-Seq method was used for gene mapping. The two bulks of normal trifoliate and multifoliolate individuals in F2population were constructed and sequenced for an average depth of 32.75´, which covered 99.22% genome compared to the reference genome. Through correlation analysis of mixed pool sequencing results by ED method, two regions were located on chromosome 11, with a total length of 5.29 Mb and a total length of 1103 genes. Three regions were identified on chromosome 11 at confidence of 0.99, with a total length of 3.42 Mb and a total of 701 genes by the association analysis of SNP-index method. There were 690 genes located simultaneously and six SNP genes between parents by the two association analysis methods. These results lay the foundation for map-based cloning of the genes related to compound leaf development. soybean; mutant; BSA-Seq 本研究由国家自然科学基金项目(31630056)资助。 This study was supported by the National Natural Science Foundation of China (31630056). 刘章雄, E-mail: liuzhangxiong@caas.cn; 邱丽娟, E-mail: qiulijuan@caas.cn E-mail: 578903659@qq.com 2020-03-24; 2020-08-19; 2020-09-02. URL: https://kns.cnki.net/kcms/detail/11.1809.S.20200902.0933.008.html 10.3724/SP.J.1006.2020.04075

2.2 测序数据分析


3 讨论
3.1 复叶发育相关基因
3.2 多小叶性状的遗传特性
3.3 候选基因的功能
4 结论
猜你喜欢
High Technology Letters(2021年4期)2022-01-09 02:08:16
河北果树(2020年4期)2020-11-26 06:05:00
海峡姐妹(2019年1期)2019-03-23 02:42:40
中成药(2018年9期)2018-10-09 07:18:46
安徽医科大学学报(2016年12期)2017-01-15 14:21:44
山东农业工程学院学报(2016年6期)2016-12-01 05:38:19
东北林业大学学报(2016年6期)2016-07-15 10:12:53
九江学院学报(自然科学版)(2015年1期)2015-11-12 03:33:20
山东医药(2015年40期)2015-02-28 14:28:45
安徽农业科学(2014年7期)2014-04-29 10:49:32
