
锌镁共掺反蛋白石羟基磷灰石的制备及其药物缓释性能
2020-11-25 04:12冯丽娜王莉丽周烨民王秀峰刘鑫鑫侯旭日
陕西科技大学学报 2020年6期
冯丽娜, 王莉丽, 周烨民, 王秀峰, 刘鑫鑫, 侯旭日
(陕西科技大学 材料科学与工程学院 陕西省无机材料绿色制备与功能化重点实验室, 陕西 西安 710021)
0 引言
近年来,骨科类疾病及相关损害的发生率迅速增加,成为对健康的重大威胁.虽然目前的给药方式能有效地预防和缓解骨病症状,但大多存在有效性低、副作用大等问题[1,2].羟基磷灰石(HA)是人体硬组织的主要成分,由于其良好的生物相容性、生物活性、骨传导性和对药物、蛋白质及DNA的高亲和力,被广泛用作药物传递、非病毒基因、抗原、酶和蛋白质的载体[3-5].
研究表明,骨中大多数天然磷灰石是非化学计量学(缺钙磷灰石晶体),由许多取代元素组成,如阳离子(Mg2+、Mn2+、Zn2+、Na+)或阴离子(HPO42-或CO32-)[6-8].在所有这些离子中,Zn2+和Mg2+是骨和结缔组织所需的微量元素,在体外和体内,通过刺激成骨细胞的增殖和胶原合成,对成骨细胞骨形成有特异性的成骨作用,对成骨细胞骨吸收有选择性的抑制作用[9].
此外,Zn2+和Mg2+可以通过改变样品性质直接或间接影响磷酸钙的生物活性,对骨骼组织的生理过程产生直接影响[10-12].考虑到Zn2+、Mg2+的重要作用,将其掺入HA可以有效地影响HA的理化性质和生物学性质[13].因此,与纯HA相比,掺杂的HA在生物材料中具有更大的应用潜力[14].
目前,许多学者对于锌和镁分别掺杂的HA在生物医用材料方面进行了大量的研究.Hyehyun Kim等[15]采用共沉淀技术合成了纯相及掺锌纳米HA作为抗癌药物阿霉素的载体并进行对比分析.结果表明,相比于纯相HA,掺锌纳米HA明显改善了对药物的负载量,提升了样品的负载能力.Dasgupta等[16]通过对纯相、掺锌及掺镁纳米HA作为蛋白质药物载体的制备并进行了体外药物缓释实验,结果表明,锌镁掺杂HA的药物释放速率均高于纯相HA,并证明锌镁的掺杂提高了HA的生物降解能力.
虽然已有大量对掺锌掺镁HA作为药物载体的研究报道,但大多都专注于单一掺杂,针对的是纳米HA的掺杂研究.本文提出将反蛋白石结构用于药物缓释来控制药物的释放速率,通过胶晶模板法制备不同掺杂量的锌镁共掺反蛋白石HA药物载体,分析了不同锌镁掺杂量对HA的形貌、物相、官能团的影响规律,进行以阿莫西林为药物模型的药物缓释性能实验,探究锌镁掺杂量对其药物缓释性能的影响规律及内在机理并将其与纯相反蛋白石HA进行了对比分析.
1 实验部分
1.1 原料
硝酸钙(Ca(NO3)2·4H2O,纯度≥99.0%,天津市风船化学试剂科技有限公司)、氯化镁(MgCl2·6H2O,纯度≥98.0%,天津市天力化学试剂有限公司)氯化锌(ZnCl2,纯度≥98.0%,天津市天力化学试剂有限公司)无水乙醇(C2H5OH,纯度≥99.7%,天津市富宇精细化工有限公司).
1.2 锌镁共掺前后反蛋白石羟基磷灰石的制备
以苯乙烯单体为原料,采用无皂乳液聚合法制备了聚苯乙烯(PS)微球,通过蒸发自组装制备了PS胶晶模板.以硝酸钙和磷酸氢二铵为钙源和磷源,保持钙与磷摩尔比为1.67.将钙源溶于乙醇溶液,磷源溶于水溶液,待完全溶解且混合后,调节溶液pH约为10,于40 ℃下恒温搅拌48 h,制备了混合分散剂体系HA前驱体.取1 g制备好的PS胶晶模板均匀平铺在外径为80 mm的布氏漏斗中.称取15 g的HA前驱体,用胶头滴管均匀地滴在PS胶晶模板上,浸渍5 min后抽滤5 min.抽滤后的样品盛放于瓷舟中,在室温下放置24 h待晾干后放入电阻炉中去除样品中的有机物微球,得到反蛋白石HA.按照氯化锌与氯化镁摩尔比1∶1取代钙,且锌镁加入量分别为1 mol%、2 mol%、3 mol%、4 mol%和5 mol%,保持钙锌镁与磷源的摩尔比为约为1.67.在其它工艺不变的情况下制备了不同掺杂量的锌镁共掺反蛋白石HA.
1.3 锌镁掺杂前后反蛋白石羟基磷灰石药物缓释性能实验
配制浓度为10 mg/mL的阿莫西林/PBS缓冲液.将上述制备的样品各取10 g浸泡在200 mL的混合液中,在室温下静置24 h后,将溶液过滤,取出固体于60 ℃烘干,烘干后的质量变化即为载药量.将载药完成的样品置于200 mL的PBS缓冲液中,保持温度为37 ℃静置,起初10 h内,在2 h、3 h、6 h和10 h分别取2 mL的上清液放入离心管内待测,同时补充相同浓度的新鲜PBS缓冲液.之后每隔5 h取一次样并加入相同浓度的PBS缓冲液直到40 h后.
1.4 样品表征
PS胶晶模板、纯相HA及锌镁共掺反蛋白石HA的微观形貌由场发射电子显微镜(S-4800,日立公司)表征.锌镁共掺前后反蛋白石HA的物相由X射线衍射仪(D/max2200PC,理学公司,日本)分析.采用Renishaw-invia型显微共焦激光拉曼光谱仪对锌镁共掺前后反蛋白石HA的官能团进行测定;不同锌镁共掺杂量及掺杂前后反蛋白石HA的载药量由分析天平测定.采用紫外分光光度计测定纯相HA及锌镁共掺反蛋白石HA的释药性能.
2 结果与讨论
2.1 锌镁共掺反蛋白石羟基磷灰石微观形貌分析
图1为单分散的PS微球和PS胶体晶体模板的SEM图.由图1(a)可知,制备的PS微球表面清洁、形貌规则、单分散性良好、平均粒径为380±10 nm.由图1(b)可知,PS胶体晶体模板排列良好,基本按照密排六方排列,此工艺下制备出了形貌完整,质量优良的PS胶晶模板.

(a)PS微球 (b)胶晶模板图1 PS微球及胶晶体模板的SEM图
图2为不同锌镁共掺杂量的反蛋白石HA的SEM图.由图2可知,不同锌镁共掺杂量下样品的微观形貌都呈现出反蛋白石结构,但是随着锌镁掺杂量的增多,样品的微观形貌中缺陷增多,结构松散并残留些许前驱体杂质.
如图2(a)所示,当锌镁共掺量均为1 mol%时,制得的样品基本呈现出良好的反蛋白石结构,少部分区域出现了未完全浸渍的前驱体杂质粘连覆盖在孔结构上.随着锌镁共掺杂量的增加,如图2(b)~(d)所示,样品中的反蛋白石结构缺陷增加,孔壁发生坍塌、变形、缺失,孔结构被堵塞且颗粒细化,未浸入模板空隙中的前驱体增多并覆盖在PS微球上,导致煅烧后团聚形成块状残留物覆盖在反蛋白石结构表面.由于锌源镁源的引入,在制备前驱体溶液时随着掺杂量增多会改变前驱体的成分,煅烧时产生其他不同相.在煅烧过程中,不同相的横向或纵向生长速度不同,导致反蛋白石结构的孔壁厚薄不同,孔壁间的结合力不同使孔壁变形、缺失.因此,当锌镁共掺杂量均为1 mol%时,得到了缺陷较少,形貌基本良好的反蛋白石HA.

(a)1 mol% (b)2 mol%

(c)3 mol% (d)4mol%

(e)5mol%图2 不同锌镁掺杂量的反蛋白石羟基磷灰石的SEM图
2.2 锌镁共掺杂前后反蛋白石羟基磷灰石微观形貌分析
图3为锌镁共掺杂前后反蛋白石HA的形貌及拉曼分析.由图3可知,锌镁共掺杂前后样品的微观形貌中都呈现出反蛋白石结构.在掺杂1 mol%锌和1 mol%镁后,样品表面有块状、棒状前驱体残留,且有较为明显的孔壁坍塌及残留物覆盖孔道的缺陷存在,如图3(a)所示.相比之下,未掺杂的反蛋白石HA显示出孔壁完整、残留物较少的良好的微观形貌,如图3(b)所示.在毛细管力和抽力作用下,前驱体溶液基本完全填充至PS模板的空隙中,因此能够将蛋白石结构完整的拓印,在煅烧后获得了孔壁厚薄均匀,孔径大小均一,结构较为完整反蛋白石HA[17].图3(c)为锌镁共掺前后样品的拉曼图.锌镁掺杂后(PO4)3-位于431 cm-1处的拉曼峰几乎消失,处于631 cm-1、1 062 cm-1处的拉曼峰发生位移且峰变得低且宽.这与Yuan等[18]的研究结果一致.

(c)拉曼图3 锌镁共掺杂前后反蛋白石羟基磷灰石的SEM及拉曼图
2.3 锌镁共掺杂前后对反蛋白石羟基磷灰石的物相分析
图4为锌镁共掺杂前后反蛋白石HA的XRD图谱.由图4(a)可知,不同锌镁掺杂量下的样品的衍射峰均与HA标准卡片(JCPDS卡号09-0432)的特征峰相对应.当锌镁掺杂量为1 mol%时,样品为较为纯净的HA.当掺杂量为2 mol%时,样品的物相发生改变,主晶相衍射峰降低且变宽,同时开始出现β-TCP相(JCPDS卡号09-0169).随着掺杂量从3 mol%增加至5 mol%,主晶相衍射峰向β-TCP相偏移量明显,次级峰降低并消失,同时出现了第三相(Ca2.589Mg0.411)(PO4)2(JCPDS卡号87-1582).当Zn、Mg掺杂入HA后,取代钙形成新的配位多面体,Zn2+、Mg2+与Ca2+之间半径差异导致晶格参数略有收缩.因此,当掺杂量较小时,煅烧后获得物相单一,但随着锌镁掺杂量的增加,结晶度明显降低.
图4(b)为锌镁共掺前后对比图,如图所示,未掺杂的反蛋白石HA衍射峰尖锐,峰值较高,结晶度较好.锌镁掺杂量为1% mol的样品中,(211)晶面的主晶相衍射峰下降,且掺杂后(511)晶面的特征峰近乎消失,掺杂后样品的结晶度降低.另一方面,Suchanek等[19]通过对锌镁掺杂量低于1% mol的HA结构研究可知,低于1% mol的掺杂对晶格及物相的影响程度较低,进而对HA理化性质的改善能力较微弱.综上,锌镁共掺杂量为1% mol时得到的样品较符合实验要求.

(a)不同锌镁共掺量

(b)掺前杂后对比图图4 锌镁共掺杂前后反蛋白石HA的XRD图
2.4 不同锌镁掺杂量的反蛋白石羟基磷灰石药物缓释性能的分析
图5为不同锌镁共掺量下的反蛋白石HA的药物缓释性能图.由图5(a)可知,不同锌镁共掺杂量的样品对阿莫西林药物负载量分别为92.144、76.236、62.809、46.921和39.556 mg/g,载药量随着共掺杂量的增加而减少.当锌镁共掺杂量为1 mol%时,样品对阿莫西林负载能力较大.由吸附质和吸附剂分子间作用力所引起的吸附称为物理吸附[20].在本实验中,药物载体羟基磷灰石与阿莫西林分子之间存在着普遍的弱吸附力,即物理吸附.一方面,由于锌镁掺杂量的增加使样品的反蛋白石结构缺陷增多,比表面积减小,物理吸附能力下降;另一方面,本实验中,阿莫西林药物分子在溶液中由于脱质子化苯酚的存在带负电荷[21],对于HA,由于氢氧根离子在材料表面的吸附,样品表面带负电,但随着锌镁掺杂量的增加样品表面的负电荷逐渐减少,这使得掺杂后的样品的与阿莫西林颗粒之间的静电力增强.两者共同作用下导致随着共掺杂的增加,载药能力下降.综上,锌镁共掺杂量为1 mol%的反蛋白石HA显示出对阿莫西林较高的负载能力.
由图5(b)可知,不同掺杂量下的样品均展示出对阿莫西林良好的缓释能力.在前5 h内,均能将药物累积释放率控制在50%内,最终累积释放率除掺杂量为1 mol%的样品外均超过90%.由于释药初期溶液中浓度差较大.因此,对阿莫西林药物的释放速率较快,之后逐渐降低.随着掺杂量的增加,药物载体与阿莫西林药物颗粒之间的静电力随着掺杂量增加而增强,导致吸附的药物颗粒难以释放.因此,锌镁共掺量为1 mol%的反蛋白石HA具有较好的药物缓释性能.
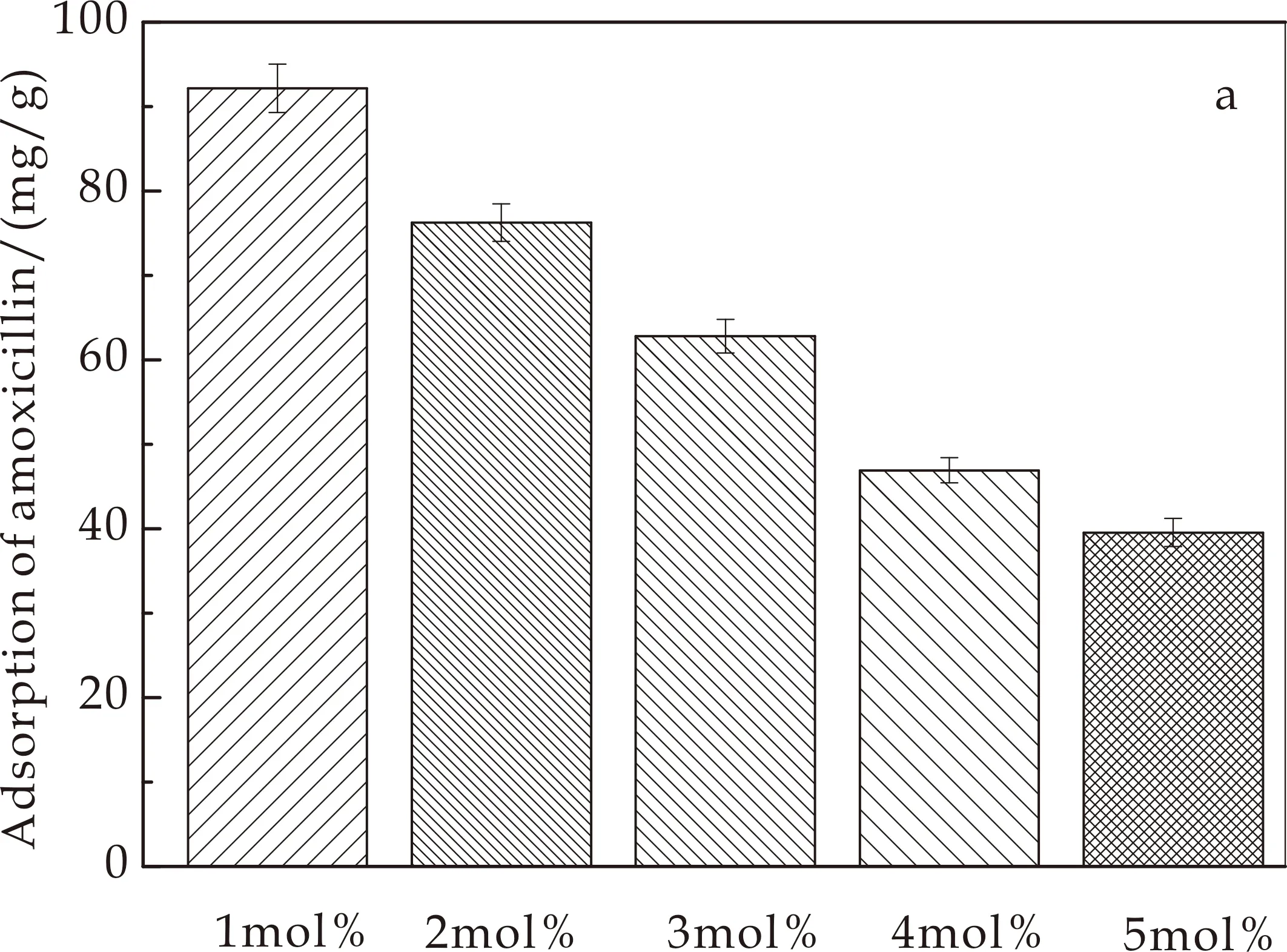
(a)载药

(b)释药图5 不同锌镁共掺杂量的反蛋白石HA的药物缓释性能图
2.5 锌镁共掺杂前后反蛋白石羟基磷灰石药物缓释性能分析
图6为锌镁共掺杂前后反蛋白石HA药物缓释性能分析.由图6(a)可知,锌镁共掺杂为1 mol%的HA载药量为92.144 mg/g,与未掺杂样品的载药量相比得到了极大的改善.由于药物分子吸附过程中主要依靠物理吸附和静电力相互作用,掺杂1 mol%后的反蛋白石HA,微观形貌中缺陷产生的较少,比表面积变化不大,故而产生的物理吸附能力的差异变化不大,吸附能力的差异主要产生于掺杂后样品与药物分子间的静电力相互作用和晶格变形而暴露的更多的活性吸附位点,锌镁粒子的掺杂均会使HA晶格畸变而产生活性吸附位点,但由于Mg2+(0.65 Å)半径小于Zn2+(0.74 Å),与Ca2+(0.99 Å)之间的畸变程度更加显著[22],因此,共掺杂中Mg2+对样品的负载能力起着主导作用.综上,锌镁共掺杂量为1 mol%的反蛋白石HA具有更好的药物负载能力.
由图6(b)可知,锌镁共掺杂前后的反蛋白石HA的药物累积释放率曲线相似,均在前5 h内将药物累积释放率控制50%,都缓解了药物的释放速率.由于锌镁的掺杂,Zn2+和Mg2+替代缺失的Ca2+后静电力相互作用增强以及微观形貌的缺陷增加使物理吸附能力降低,共同影响下使得掺杂前后药物的释放能力差异较小.因此,锌镁共掺杂前后的反蛋白石HA的药物释放能力无明显差异.

(a)载药

(b)释药图6 锌镁共掺杂前后的反蛋白石HA的药物缓释性能图
3 结论
(1)通过调节前驱体溶液中Zn2+、Mg2+的含量,采用胶晶模板法成功制备了不同锌镁掺杂量(1%~5% mol)的反蛋白石HA.
(2)随着锌镁共掺杂量的增加,样品微观形貌中缺陷增加,结晶度下降并出现杂相.锌镁共掺杂量为1mol%时的反蛋白石HA有较好的微观形貌及结晶度,显示出对阿莫西林良好的载药性能.
(3)与未掺杂相比,当锌镁共掺杂量为1 mol%时,由于掺杂后的晶格变形和缺陷增加,样品暴露了更多活性吸附位点,掺杂后的有序多孔HA对阿莫西林的载药能力得到明显改善,对于该药物的释放能力,掺杂前后未发现有明显改善.
猜你喜欢
宝藏(2022年8期)2022-09-27
现代矿业(2022年6期)2022-07-13
中国典型病例大全(2022年13期)2022-05-10
有色金属科学与工程(2022年1期)2022-03-12
家庭医药(2021年14期)2021-12-02
家庭医药·快乐养生(2021年7期)2021-07-19
昆明医科大学学报(2021年1期)2021-02-07
家庭科学·新健康(2020年6期)2020-07-06
大连工业大学学报(2020年1期)2020-01-17
武汉工程大学学报(2019年5期)2019-11-02
